BACKGROUND Diabetic foot ulcers are a common diabetic complication leading to alarming figures of amputation, disability, and early mortality. The diabetic glucooxidative environment impairs the healing response, promoting the onset of a ‘wound chronicity phenotype’. In 50% of ulcers, these non-healing wounds act as an open door for developing infections, a process facilitated by diabetic patients’ dysimmunity. Infection can elicit biofilm formation that worsens wound prognosis. How this microorganism community is able to take advantage of underlying diabetic conditions and thrive both within the wound and the diabetic host is an expanding research field.
OBJECTIVES 1) Offer an overview of the major cellular and molecular derangements of the diabetic healing process versus physiological cascades in a non-diabetic host. 2) Describe the main immunopathological aspects of diabetics’ immune response and explore how these contribute to wound infection susceptibility. 3) Conceptualize infection and biofilim in diabetic foot ulcers and analyze their dynamic interactions with wound bed cells and matrices, and their systemic effects at the organism level. 4) Offer an integrative conceptual framework of wound–dysimmunity–infection–organism damage.
EVIDENCE AQUISITION We retrieved 683 articles indexed in Medline/PubMed, SciELO, Bioline International and Google Scholar. 280 articles were selected for discussion under four major subheadings: 1) normal healing processes, 2) impaired healing processes in the diabetic population, 3) diabetic dysimmunity and 4) diabetic foot infection and its interaction with the host.
DEVELOPMENT The diabetic healing response is heterogeneous, torpid and asynchronous, leading to wound chronicity. The accumulation of senescent cells and a protracted inflammatory profile with a pro-catabolic balance hinder the proliferative response and delay re-epithelialization. Diabetes reduces the immune system’s abilities to orchestrate an appropriate antimicrobial response and offers ideal conditions for microbiota establishment and biofilm formation. Biofilm–microbial entrenchment hinders antimicrobial therapy effectiveness, amplifies the host’s pre-existing immunodepression, arrests the wound’s proliferative phase, increases localized catabolism, prolongs pathogenic inflammation and perpetuates wound chronicity. In such circumstances the infected wound may act as a proinflammatory and pro-oxidant organ superimposed onto the host, which eventually intensifies peripheral insulin resistance and disrupts homeostasis.
CONCLUSIONS The number of lower-limb amputations remains high worldwide despite continued research efforts on diabetic foot ulcers. Identifying and manipulating the molecular drivers underlying diabetic wound healing failure, and dysimmunity-driven susceptibility to infection will offer more effective therapeutic tools for the diabetic population.
ABSTRACT BACKGROUNDDiabetic foot ulcers are a common diabetic complication leading to alarming figures of amputation, disability, and early mortality. The diabetic glucooxidative environment impairs the healing response, promoting the onset of a ‘wound chronicity phenotype’. In 50% of ulcers, these non-healing wounds act as an open door for developing infections, a process facilitated by diabetic patients’ dysimmunity. Infection can elicit biofilm formation that worsens wound prognosis. How this microorganism community is able to take advantage of underlying diabetic conditions and thrive both within the wound and the diabetic host is an expanding research field.
OBJECTIVES1) Offer an overview of the major cellular and molecular derangements of the diabetic healing process versus physiological cascades in a non-diabetic host. 2) Describe the main immunopathological aspects of diabetics’ immune response and explore how these contribute to wound infection susceptibility. 3) Conceptualize infection and biofilim in diabetic foot ulcers and analyze their dynamic interactions with wound bed cells and matrices, and their systemic effects at the organism level. 4) Offer an integrative conceptual framework of wound–dysimmunity–infection–organism damage.
EVIDENCE AQUISITION We retrieved 683 articles indexed in Medline/PubMed, SciELO, Bioline International and Google Scholar. 280 articles were selected for discussion under four major subheadings: 1) normal healing processes, 2) impaired healing processes in the diabetic population, 3) diabetic dysimmunity and 4) diabetic foot infection and its interaction with the host.
DEVELOPMENTThe diabetic healing response is heterogeneous, torpid and asynchronous, leading to wound chronicity. The accumulation of senescent cells and a protracted inflammatory profile with a pro-catabolic balance hinder the proliferative response and delay re-epithelialization. Diabetes reduces the immune system’s abilities to orchestrate an appropriate antimicrobial response and offers ideal conditions for microbiota establishment and biofilm formation. Biofilm–microbial entrenchment hinders antimicrobial therapy effectiveness, amplifies the host’s pre-existing immunodepression, arrests the wound’s proliferative phase, increases localized catabolism, prolongs pathogenic inflammation and perpetuates wound chronicity. In such circumstances the infected wound may act as a proinflammatory and pro-oxidant organ superimposed onto the host, which eventually intensifies peripheral insulin resistance and disrupts homeostasis.
CONCLUSIONSThe number of lower-limb amputations remains high worldwide despite continued research efforts on diabetic foot ulcers. Identifying and manipulating the molecular drivers underlying diabetic wound healing failure, and dysimmunity-driven susceptibility to infection will offer more effective therapeutic tools for the diabetic population.
INTRODUCTION Diabetes mellitus (DM) is characterized by the onset and progression of a constellation of multi-organ complications resulting from multifactorial interactions—including biochemical derangements and epigenetic factors—which ultimately translates to irreversible tissue changes as a response to glucooxidative processes.[1] Of all diabetic complications, the development of diabetic foot ulcers (DFUs) is among the most common and debilitating.[2,3] Classic concepts define DFU as deep tissue damage of the lower limb, frequently preconditioned by, and associated with, neuropathy or peripheral arterial disease.[4] It is recognized as a major and growing public health problem, a scientific challenge and a socioeconomic burden;[5] and remains the main causal factor of lower extremity amputations, disability and early mortality.[6] Armstrong introduced the ‘cancer analogy’ concept to highlight the fact that five-year mortality rates associated with foot ulceration and amputation surpass those registered for common cancers.[7–10]
IMPORTANCE This article contrasts wound healing processes in healthy individuals and diabetics, establishing and conceptualizing the reciprocal links between diabetic dysimmunity, susceptibility to infection, diabetic foot ulcer chronicity and insulin resistance amplification.
Diabetic glucooxidative stress impairs the healing response and disrupts the flow of overlapping healing phases, ultimately promoting the onset of a ‘wound chronicity phenotype’.[11–13] Aside from healing impairment, a common occurrence in diabetic patients is ulcer recurrence after primary closure.[6] These non-healing wounds are a major predisposing factor or entry point for wound infection[11] and accordingly, more than 50% of DFUs become infected.[14] Infection acts as a primary deterrent to physiological healing responses[15] and a risk factor for lower-limb amputation,[16–18] especially when deep tissues and bones are compromised.[19,20] Although diabetics are particularly vulnerable to bacterial infections,[21–23] DFUs have a complex and highly organized polymicrobial community that frequently contributes to undesirable outcomes in DFU-affected individuals.[22] This microbiota–biofilm comprises symbiotic bacteria, yeast and fungal loads and can silently spread, amplify the underlying healing deficit, increase antibiotic resistance, disrupt host metabolism and further dampen immune response.[24–27]
Globally speaking, DM and infection increasingly go together.[28,29] Diabetic individuals are prone to peripheral-tissue infections; given dysregulations in primary surveillance, recognition, activation and neutralization mechanisms within the innate immunity repertoire.[21,30,31] Furthermore, diabetic individuals exhibit antigen presentation failure, contraction of T-cell–mediated immune function[32] and a particular predisposition to bacterial adhesion to epithelial linings.[33,34]
DFU is a unique battlefield where host–microorganism interactions shape ulcer progression. Consequently, numerous studies have addressed the role of biofilm on DFU and its impact within the ulcer bed and the host itself.[35–38] We reviewed this critical issue, given its etiopathogenic relevance to basic aspects of DFU pathology: 1) Why are diabetic persons more susceptible to wound infections? 2) How is DFU biofilm organized? 3) How does a microorganism’s pathogenic potential and concentration impact DFU outcomes? and 4) How does biofilm impair the healing response?
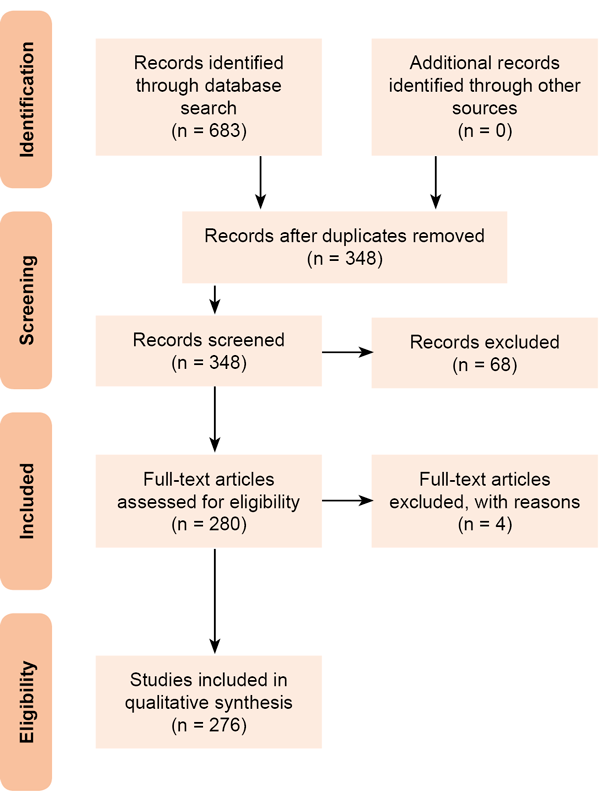
EVIDENCE ACQUISITION We retrieved articles indexed in Medline/PubMed, SciELO, Bioline International, and Google Scholar using the following keywords/phrases: DFU, limb AND amputation, DFU AND infection, DFU AND biofilm, immune system AND diabetic patient, microorganism AND immune system. A total of 683 articles were retrieved and exported a reference manager. Duplicate articles were removed (Figure 1). Our final selection included 280 research and review articles. Titles, objectives and abstracts were carefully screened and reviewed. The search was limited to the English language without date restrictions. All compiled information was structured under four principal headings: 1) a general overview of the normal healing response in a healthy organism; 2) an overview of the cellular and molecular foundations of the impaired healing process in the diabetic population; 3) diabetic dysimmunity; and 4) conceptual definition and pathogenic implications of diabetic foot infection (DFI) in its interaction with the host.
Figure 1. Literature review process
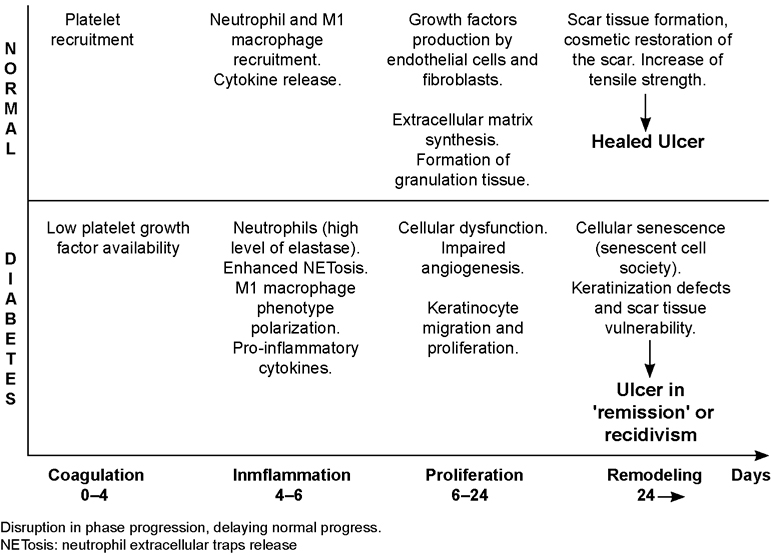
DEVELOPMENT Brief overview of normal healing response Wound healing is a dynamic and complex process that ultimately results in restoration of anatomic integrity with analogous function.[39–41] Of note, however, skin wound healing represents an evolutionary advantage for organism survival, given its role in restoring barrier function, as well as preventing internal tissue damage and infection dissemination.[42] This evolutionary advantage involves a complex and intricate, but finely regulated, crosstalk between cells and soluble mediators.[43] A normal healing process is made up by four overlapping phases: 1) coagulative, 2) inflammatory, 3) proliferative, and 4) remodeling (Figure 2). Each phase takes place during a temporary window involving a certain cell population, a specific set of cytokines and a particular chemical composition within the extracellular matrix (ECM).[44,45] The coagulation process, aside from ensuring hemostasis, has two other relevant functions: 1) the fibrin clot and fibrinogen byproducts act as a scaffold and chemoattractant for the recruitment and anchorage of inflammatory cells, fibroblasts and other mesenchymal-derived cells that will participate in tissue granulation formation; and 2) platelet degranulation promotes primary growth factors. Platelets represent the first group of resident cells with fibroangiogenic soluble messengers, including platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), epidermal growth factor (EGF) and insulin-like growth factor-1 (IGF-1).[46,47]
Of particular relevance for healing trajectory and for the ultimate scar phenotype are the infiltrating inflammatory cells and their biochemical signalers, as they exist in interaction with granulation tissue-resident cells as fibroblasts, myofibroblasts and endothelial cells.[44] Since cutaneous injury is linked to the release of ‘danger and pathogen signals’, innate immune receptors become activated and eventually trigger an inflammatory phase.[48]
The influx of polymorphonuclear neutrophils (PMNs) is ensured mainly by a complex cascade of vasoactive signalers and chemoattractants, so that these cells invade and are anchored within the wound matrix, initiating the wound’s acute phase that may last up to four days.[49] PMNs pattern recognition receptors are activated by local damage-associated molecular patterns (DAMP) released during cell injury and necrosis, and by bacterial pathogens’ associated molecular pattern (PAMP). Upon activation, these cells release pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β) and IL-6, which amplify the inflammatory response and pave the way for macrophage infiltration and activation.[50] Activated PMNs ‘clean’ the wound bed of tissue debris via an armamentarium of degradative and antimicrobial proteases (cathepsins, defensins, lactoferrin and lysosomes) stored in cytoplasmic granules. This sanitizing process is largely dependent on the formation of neutrophil extracellular traps (NETs), web-like structures that capture and eliminate exogenous bacteria, fungi and viruses.[51] This process of NET release is termed NETosis and has broad implications for different forms of inflammation.[52] Recent studies show that circulating PMNs from diabetic subjects are biased toward excessive release of NETs,[53] and that uncontrolled NETosis impairs the healing process in diabetic mice and humans.[54] These cells also generate reactive oxygen species (ROS) that help eliminate invading pathogens.[55] Altogether, the role of PMNs in wound healing is undoubtedly important, but an extension of their local residence time and their functional profile may lead to wound chronicity (Figure 2).
Figure 2. Normal vs. diabetic healing
A second generation of inflammatory infiltrating cells comes from local resident macrophages that are differentiated from infiltrating monocytes, which are activated via DAMP, PAMP and a cytokine surge.[56] Anti-macrophage depletion studies have revealed the physiological significance of these cells for the late stage of the inflammatory phase and for initiation of the proliferative one.[41,57,58] This late stage of the inflammatory phase is mostly characterized by the M1 subset of macrophages, which themselves are characterized by phagocytic and pro-inflammatory activities. Later, macrophages polarize to an M2 subpopulation with anti-inflammatory activity by expressing interleukin-receptor antagonists and a collection of fibroangiogenic growth factors that enhance fibroblast proliferation, extracellular matrix synthesis, angiogenesis,[59,60] ultimately reducing inflammation.[47] Macrophage subclass shift from M1 to M2 subsets is a meaningful event, given its role in turning off inflammation, clearing the wound bed of apoptotic PMNs (efferocytosis), ensuring proliferative phase progression, and preventing autoreactivity to released self-antigens.[15,57,61] Conclusively, inflammation is a time-restricted, finely controlled sequential event, whose expansion is paradigmatically associated with a torpid healing phenotype, or to wound chronification and senescence of granulation tissue productive cells.[16,17,62]
Granulation tissue is subsequently organized and populated by a broad spectrum of extracellular matrices, secreting cells that are in active and dynamic engagement with the substrate, and progressively modulating the structure and composition of the wound’s extracellular matrix.[63] Although granulation tissue is a temporary organ, it is important as a ‘welding material’ filling wound gaps, preventing environmental threats, and providing support for cell adhesion, migration, growth and differentiation during wound repair.[63,64]
The proliferative phase also embraces three important processes: angiogenesis, wound contraction and re-epithelialization.
Angiogenesis is an exciting biological process actively regulated by pro-angiogenic growth factors, chemokines, integrin receptors, bone marrow-derived progenitor cells, and transcriptional and post-transcriptional epigenetic regulators; it restores blood inflow and outflow, and therefore oxygen delivery and CO2 extraction. Its role in a normal healing trajectory is essential.[65] In response to pro-angiogenic signals like vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), PDGF-B, TGF-β, and angiopoietins, endothelial cells initiate angiogenesis by sprouting, proliferation and migration.[66] At the same time, locally secreted antiangiogenic factors are able to counterbalance and limit excessive angiogenesis.[67] The lack of an appropriate angiogenic response is a representative hallmark of DFUs, and identification of molecular forces underlying diabetic microangiopathy has been extensively examined.[68,69]
Wound contraction and epithelial resurfacing are two integrated mechanisms that ensure complete wound closure in most mammalian species. Contraction, a physiological and necessary event in wound closure, appears to be mediated by myofibroblasts, specialized cells responsible for force generation, that are differentiated from migrating fibroblasts during granulation tissue formation.[70] Although current theories posit that alpha smooth muscle actin-expressing myofibroblasts contribute to wound contraction,[71] others attest that contraction progresses through fibroblast-derived traction forces via thick collagen fiber secretion.[72] Irrespective of the contraction-responsible cell and the molecular drivers behind them, limited contraction is associated with torpid healing in diabetic wounds.[12]
Instrumental for complete and successful wound closure is re-epithelialization. This event demands integration of leading-edge keratinocyte proliferation, migration and differentiation in order to re-establish epidermal integrity.[73] This is perhaps one of the most complex and unexplored processes in wound healing.[74–76] Simplistically described, keratinocytes at the wound edge and epithelial cells from hair follicles in the vicinity migrate and proliferate. Signals that promote keratinocyte proliferation include heparin-binding EGF-like growth factor (HB-EGF), EGF, transforming growth factor alpha (TGF-α), and FGF secreted from platelets, macrophages and dermal fibroblasts.[77] For migration to progress, there is a reduction of desmosomes and hemidesmosome connection, cytoskeleton reorganization, morphological reprograming and changes in the pattern of keratin expression.[78] In general, there is a loss of physical tension at points of cell attachment to the basal lamina.
Re-epithelialization progresses when the basement membrane is reconstituted by upper dermal fibroblasts and keratinocytes in a cooperative effort.[79] In this context, the interaction between integrin receptors and the neomatrix determines the speed of migrating keratinocytes, a process that in turn is regulated by growth factor gradients.[80] Also important are plasmin and other plasmin-related degradative enzymes that degrade fibrin and other matrix glycoproteins, facilitating keratinocyte migration. Other collagenases and gelatinases are expressed by migrating keratinocytes.[77] Once keratinocytes attach to the basement membrane, they initiate a process of upward migration and differentiation to create a mature stratified, squamous epithelium that covers the wound.[81] Failed or delayed re-epithelialization is an obvious sign of wound stagnancy.[82] Compelling evidence documents the deleterious role of hyperglycemia and other downstream biochemical signalers on fibroblast and keratinocyte proliferation and migration.[83–85]
The ultimate phase—remodeling—begins approximately 14–21 days post-injury and may continue for years.[86–88] Its main function is the formation of normal epithelia and maturation of scar tissue;[89] excessive matrix is eliminated by a set of metalloproteinases, while new type-1 collagen fibers are produced and horizontally aligned in a more organized and esthetic manner. Inflammatory infiltration has ceased and different cell populations gradually enter into apoptosis.[90] The initial granulation tissue progressively collapses and is replaced by a new wound chemical milieu, so that the scar ECM architecture increasingly approaches original tissue morphology. The complexity involved in wound tissue remodeling has led us to hypothesize the existence of structural, positional and organizational memory in late wound cells, enforced by topographic ‘home address signals’. At this point, wound tensile strength is restored, and antiangiogenic mediators are locally released to turn off angiogenic sprouting and ensure excessive vessel regression. In parallel, other anatomical structures including the epidermis, nerves and myofibers are synchronously remodeled forming a functional unit.[78,86,91]
The diabetic foot ulcer: overview of the molecular bases of the torpid healing process Although it is generally accepted that time-to-heal determines a wound’s clinical classification as acute or chronic, the conceptual definition of wound chronicity has remained controversial.[92] Nevertheless, it is generally accepted that a wound is considered chronic when it fails to proceed in “an orderly and timely reparative process that results in sustained restoration of anatomic and functional integrity”.[92] DFU is archetypical of chronic wounds.[93–95] It is generally accepted that a primary hallmark of diabetic wounds is their persistent arrest in an unproductive inflammatory phase, associated with impaired formation and consolidation of mature granulation tissue.[96,97]
The arrest in this inflammatory phase is not associated with successful control of local infection, and thus it has been proposed that diabetic individuals are more vulnerable to wound infection[98,99] due to the existence of a primary deficit in innate immune response mechanisms.[100,101]
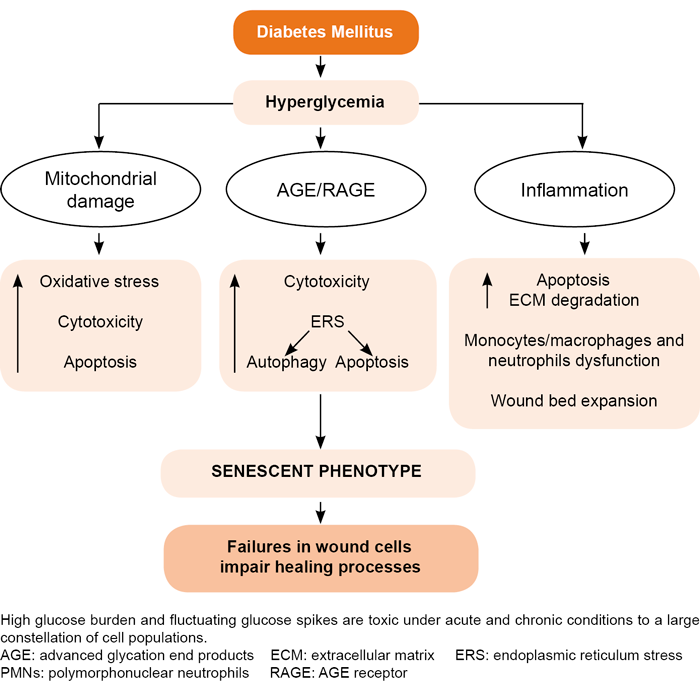
Hyperglycemia, again, seems to act as the proximal trigger for an exaggerated inflammatory reaction (Figure 3). Hyperglycemia and its distal operators—advanced glycation end products (AGE), TNF-α and other pro-inflammatory signalers—exert profound cytotoxic effects in fundamental ‘building-block’ cells of describes tissue.[11,102] Compelling evidence describes an inflammation-prone, pro-oxidative and pro-degradative environment in the core of diabetic wounds.[103–106]
Uncontrolled pro-inflammatory cytokine secretion imposes a pro-catabolic balance in the wound bed that both increases peripheral insulin resistance and reduces injured tissue’s anabolic response.[107,108] TNF-α downregulates fibroblast collagen synthesis in diabetic skin and upregulates the synthesis of metalloproteinases by amplifying the wound’s proteolytic and pro-degradative profile.[109,110] Although some studies have ruled out hyper- or hypoglycemia’s role in significantly disrupting PMNs cells’ ability to enter into apoptosis,[111] others have pointed to poorly controlled glycemia levels as a major factor in the prolonged residence of apoptosis-resistant PMNs with active secretory functions,[111] which ultimately translates into an elevated proteolytic/degradative balance.[112,113] This pro-degradative environment reduces local availability of growth factors and their receptors, hindering the ability of fibroblasts and endothelial cells to participate fully in the healing process.[88,114] PMN are also considered a source of ROS and nitric oxide species within the wound bed, with remarkable cytotoxic and pro-degradative potential.[115,116] The increased rate of ROS is an indirect consequence of poorly-controlled glucose levels given that existing evidence provides a connection between the high-glucose–induced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase pathway and exaggerated NETosis.[117] Hyperglycemia also induces myeloid progenitor proliferation and expansion, as well as increased neutrophil production of S100A8/S100A9, which ultimately binds to AGE receptors (RAGE). This interaction translates to enhanced ROS production and myelopoiesis[118] that reinforces the triad of hyperglycemia, neutrophil infiltration and local ROS production.
Aside from PMN, M1 subclass macrophages predominate in diabetic wounds.[119] Although the molecular drivers behind this process are not yet fully understood, it has been demonstrated that human monocytes and macrophages undergo M1-like inflammatory polarization when exposed to high levels of glucose in both culture conditions and in hyperglycemic subjects.[120] This hyperpolarization to the pro-inflammatory arm represents a flaw in the transition to the M2 subclass and, therefore, to an anti-inflammatory and pro-healing profile. Additionally, diabetic wound macrophages exhibit defective efferocytosis, a mechanism for clearing apoptotic bodies in the wound bed.[121] This failure increases the local surge of proinflammatory cytokines, which perpetuates inflammation and increases the risk of it becoming chronic.[121] Another study has documented that hyperglycemia itself, without additional metabolic factors, induces a mixed profile of M1/M2 cytokines that nurture diabetes-associated inflammation and atherosclerosis.[122] The diabetic systemic low-grade inflammatory phenotype is able to introduce monocyte chromatin modification that, in turn, intensifies the persistent pro-inflammatory state.[123]
The proliferative phase in diabetic foot ulcer healing is frequently slow, torpid and asynchronous.[124] This may entail irregular and abnormal fibroblast recruitment, scarce or abnormal extracellular matrix protein secretion, limited cell-anchoring scaffold synthesis, poor or abnormal angiogenesis—including pathologic vascular remodeling, slow contraction of wound contours, torpid re-epithelialization and the inability to remain in remission after epithelial resurfacing.[11,125,126] From the molecular angle, compelling evidence has identified high glucose burdens and accompanying fluctuating glycemia spikes[127] as the proximal trigger of many cellular impairments that generically transform into fibroblasts, endothelial cells and keratinocytes in mitogenic and motogenic arrest; premature apoptosis and the onset of a senescent phenotype.[128–130] Multiple de novo circuitries, metabolic shunts, inflammatory-prone reactants and abnormal pathways in diabetics impact the wound-healing response and perpetuate the ulcer.[131,132]
Glycoxidation derivatives are intrinsically cytotoxic to productive cells in granulation tissue, and further amplify pro-inflammatory and pro-oxidative circuitry by binding to RAGE.[133,134] Glycoxidative products accumulate in non-labile dermal collagen[132,134] leading to cutaneous cell toxicity and premature senescence, impairing fibroblast and endothelial cell physiology,[99] and consequently delaying granulation tissue formation and maturation.[11,133] Conclusively, within the wound, the triad of TNF-α, ROS and AGE can initiate apoptosis of fibroblasts and vascular cells, thereby prolonging inflammation, reducing growth factor availability and opening the gate for the onset of the so-called ‘wound senescent cell society’.[130,135–137] It is not surprising therefore, that repair-committed cells in diabetics move through proliferative arrest, senescence, and apoptosis (Figure 3).[136]
Re-epithelialization failure in diabetics and the tendency toward local recidivism are significant challenges for clinicians, wound care providers, and basic scientists. It has been suggested that an incomplete program of keratinocyte activation and differentiation[138] is fundamental for the presence of mitotically active—but not migrating—epithelial cells along the wound’s leading edge.[73,76] High glucose has shown to exhibit a toxic effect on keratinocytes, reducing proliferation, replicative life span,[139] and migratory responses.[140] The fact is that, as long as the wound is not resurfaced, the threat of infection and amputation remains.
Diabetes and infection susceptibility The relationship between DM type 2 (DMT2) and immunity is an expanding research field in which new puzzle pieces are continuously discovered, often increasing in complexity and stimulating controversy within the field. This research incentive is fueled by the understanding that diabetes increases the risk of certain infections[141] and infection-related mortality.[142]
DMT2 is currently considered an immunometabolic disease, given the role of T-lymphocyte activation in inflammation and in the onset of insulin resistance.[143] The robust pathogenic loops linking insulin secretion, peripheral insulin resistance, immuno-inflammation and DMT2 are beyond the scope of this analysis, and have already been thoroughly reviewed.[144–146] The links are well defined: inflammation leads to peripheral insulin resistance and, in turn, insulin resistance leads to inflammation.[147] However, it is important to note that inflammation does not necessarily represent immunocompetence. On the contrary, some experimental data suggest that diabetes-associated hyperinflammation amplifies damage from bacterial infections and leads to increased susceptibility to Gram-negative bacteria.[148] Since the actual cause of death of the mice in one study was hyperinflammation, the authors suggest that this rather counterintuitive finding may respond to diabetes-associated RAGE overexpression, which preconditions a chronic inflammatory scenario that ultimately may be lethal when presented with the additional challenge of a bacterial infection.[138] Cumulative evidence documents a significant correlation between infection and rate of glycemic control.[21,149,150] It follows that heightened susceptibility to infections would be associated with insufficient glycemic control.[151,152] This observation is particularly relevant in the scope of our review to cellulitis, DFU, the devastating conditions of necrotizing fasciitis, and Fournier’s gangrene.[22]
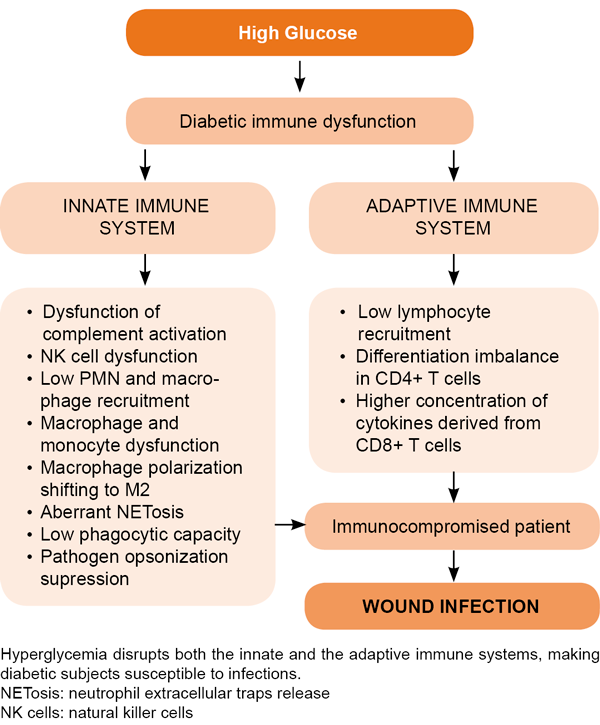
Hyperglycemia and insulin deficiency are considered the two major etiopathogenic pillars of diabetes-associated immunodeficiency (Figure 4) and susceptibility to infections.[153–155] Hyperglycemia as a primary trigger of pro-inflammatory cytokine spillover[145] results in local and systemic inflammation, and peripheral insulin resistance.[156,157] Increased levels of various inflammatory markers and mediators—including white blood cell count, C-reactive protein, pro-inflammatory cytokines and plasminogen activator inhibitor-1—are elevated in insulin resistant subjects and DMT2-affected patients.[158,159] Although it may appear contradictory, evidence suggests that peripheral blood mononuclear cells (PBMC) from DMT1 and DMT2 patients secrete lower constitutive-[160] and lipopolysaccharide (LPS)-stimulated[161] levels of TNF-α, IL-1 (α and β) and IL-6, as compared with matched controls. The same cytokine secretion impairment was confirmed for in vitro models where PBMCs from healthy donors were exposed to high glucose levels[162] or dextrose octreotide.[163] This study demonstrates that glucose exposure dampened IL-2, IL-6 and IL-10 levels in a concentration-dependent manner while conversely inducing the expression of TGF-β1 which may explain immune failure.[162]
Increased glycation leads to a reduction of IL-10 secretion by myeloid cells.[32] A recent study demonstrated that PBMC steady-state expression of IL-1β appeared significantly increased while IL-6 expression reduced 3.45-fold in a cohort of DMT2 patients, compared with healthy control subjects.[164] This is a counterintuitive observation considering the canonic links between IL-6, glucose metabolism, DMT2 and their complications.[165–168]
Aside from these findings, a wealth of classic studies associated elevated endovascular levels of pro-inflammatory cytokines with insulin resistance and DMT2.[169,170] Subclinical blood elevation of some of these markers anticipates the onset of DMT2,[171,172] and the progression of multi-organ complications.[173,174] More recent studies[175] have identified and expanded epigenetic explanations as to why hyperglycemia induces long-lasting inflammatory upheaval, even days after glucose normalization. The fact is that hyperglycemia-associated chronic inflammation is epigenetically sculpted and is one of the biochemical insignias of metabolic memory.[176] Therefore, unraveling the roots of this plethora of conceptual controversies requires additional studies.[177–179]
Incorporation of AGEs to non-diabetic–derived cells has conclusively shown elevations in cytokine secretion,[100] which led to the hypothesis that AGE is somehow involved in diabetics’ increased basal cytokine secretion.[100] Glycation also reduces expression of class-I major histocompatibility complex on the surface of myeloid cells, which accounts for impaired cellular immunity.[32] The presence of high glucose concentrations leads to elevated constitutive (steady state) cytokine production in resting cells, which becomes insufficient upon PBMC stimulation. Cell stimulation provoked by cytokine insufficiency is thought to be a critical factor in impaired immunity and vulnerability to invading pathogens among diabetics.[32,162,163,180] However, an alternative line of thought has suggested that stimulating the immune system will imperil—rather than protect—diabetics from acute Gram-negative bacterial infections, and that dampening hyperinflammation (with dexamethasone, for example) may restore the innate immune response to such infections.[148]
Figure 4. Diabetes impairs immune response and predisposes patients to wound infection
Other studies have addressed leukocyte recruitment and pathogen recognition abilities. Animal models demonstrate that diabetes impairs CD45+ leukocyte and CD8+ T cell recruitment, and reduces adhesion molecule expression and cytokine production upon microbial invasion of different organs.[181,182] More conflicting evidence exists, however, regarding pathogen recognition via toll-like receptors (TLR). One line indicates that TLR expression is reduced in diabetic mice.[182,183] In contrast, studies in human samples attest that TLR expression is low in diabetic subjects with complications and deficient glycemic control, whereas it is high in patients with well-controlled glycemia without complications.[184] Overall it has been concluded that TLR pathway impairments lead to diminished recognition of bacteria and may be one of the mechanisms implicated in diabetic susceptibility to wound infection.[183]
More coincidental revelations stem from the characterization of neutrophil dysfunction documented in diabetic individuals or in healthy donors exposed to high glucose burdens. The so-called ‘oxidative burst’ plays a critical role in neutrophils’ antimicrobial defense and is reduced upon high glucose concentration due to poor ROS and superoxide anion productions.[185,186] Other studies have documented that sustained hyperglycemia leads to neutrophils’ functional decline and that the mechanisms behind this decline include increased adhesive capacity, as well as diminished chemotaxis, phagocytic activity and bactericidal capacity.[102,187,188] This is a conflicting observation given the solid evidence supporting the concept that hyperglycemia is a major trigger of inflammation and hyper-oxidation in diabetes.[189–191]
Other studies show lower neutrophil degranulation,[192] decreased phagocytic capabilities,[193] immunoglobulin-mediated opsonization inhibition[194] and limited NET response ability.[195] These conflicts could be due to the type and origin of the experimental designs of, and samples used in, the studies; however, the overall interpretation of these data has led to the conclusion that hyperglycemia dampens PMN chemotaxis and phagocytic activities.[21] Diabetes impairs the physiology of macrophages and other innate immune cells. As stated above, diabetic patients are highly susceptible to bacterial infections, and often have impaired wound healing. However, despite years of research, the molecular mechanism underlying macrophage dysfunction in diabetes is not fully understood .[196]
Under high glucose conditions, in vitro and in vivo models have shown reduced complement receptors, adhesion capacity, phagocytosis and antibacterial activity.[197] Diabetic hyperlipidemia and hyperglycemia introduce epigenetic modifications to macrophages that promote the onset of an inflammatory phenotype.[198] Thus, high glucose levels induce inflammatory polarization of human macrophages in vitro whereas AGEs significantly prime and promote M1 macrophage markers expression and IL-6 and TNF-α secretion.[199] Accordingly, M1 macrophages have strong microbicidal and antigen-presenting capacities, produce pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), and ROS; whereas M2 macrophages are considered pro-resolution response cells, producing anti-inflammatory mediators (IL-10 and TGF-β) and consequently resolving inflammation.[200] In light of these observations, it is controversial to posit M2 macrophage polarization in diabetic mice (db/db) as an explanation for their susceptibility to bacterial infection.[196] As a matter of fact, M2 cells are scarce within the wound environment where they are obviously necessary.[201] Irrespective of the controversial issue of in-wound recruited macrophage polarization, a recent study confirms that in diabetic mice, macrophage phagocytosis and bactericidal activities are reduced upon long-term exposure to high glucose burdens.[196] For these authors, long-term high-glucose treatment reduced macrophage glycolytic capacity and glycolytic reserve, in turn, impairing phagocytic capability.
NK (natural killer) cells derived from diabetic individuals also demonstrate defects in activating NKG2D and NKp46 receptors related to NK degranulation failure.[196] Hyperglycemia is also associated with a reduction in C4-fragment opsonization, which inhibits classical or lectin pathways of complement activation,[202] in impaired neutrophils’ bacterial killing capacity.[100,203] By using peripheral blood lymphocytes from diabetic animal models and human samples, studies have concluded that uncontrolled diabetes increases chromatin condensation, DNA fragmentation and lymphocyte death.[204]
Unlike the effect of hyperglycemia on immune cell activity in DMT2, the impact of insulin deficiency in DMT2 immunoresponsive cells against pathogens has not been widely studied.[170] Given that these cell functions are energy-dependent processes, proper insulin-regulated glucose metabolism is necessary. Insulin-driven metabolic processes are not merely associated with immune-cell ATP generation and utilization. Glucose and lipid metabolism influences cellular phenotype, potential cellular reprograming during patterns of recognition, and ultimately activation status.[205] In activated T-lymphocytes, insulin stimulates glucose uptake, oxidation, pyruvate flux and pyruvate dehydrogenase activity, amino acid transport, lipid metabolism and protein synthesis.[206] Recent findings implicate insulin in shaping the immune response by modulating cell differentiation and polarization.[207,208] Thus, in addition to its role in substrate metabolism, insulin is also an anti-inflammatory and immunomodulatory hormone[209,210] via immune cells’ metabolic regulation.[211]
Recent studies substantiate the importance of insulin in normal innate immune response. An insulin deficit is associated with alveolar macrophages’ phagocytic impairment as well as poor cytokine secretion in alloxan-treated rats, reverted after insulin intervention.[212] Insulin treatment of diabetic mice bone marrow-derived macrophages restored production of critical pro-inflammatory cytokines upon LPS exposure.[213]
Conclusively, the apparently ‘trivial’ blood glucose derangements in diabetes reduce bactericidal and wound healing capacities in innate immune operators, a phenomenon that, according to latest evidence, is related to transcriptional aberrations in gene coding for macrophage differentiation and lymphocyte migration and proliferation at the hematopoietic stem/progenitor cell level.[214] Thus, therapeutic manipulation of immune-metabolomic loops is a promising therapeutic road.
Infection of diabetic foot ulcers: biofilm and its interaction with the wound matrix Typically, once an ulcer develops, it is colonized with microorganisms that may lead to a state of clinical infection.[113] About 15%–25% of DM patients develop foot ulcers during their lifetime and half of these become infected, a recurrent complication in diabetics.[215,216] Infection can spread to soft tissues and bone making it the main causal factor of lower extremity amputation in most countries.[217] Thus, the management of DFI represents a high cost for the health system, a decrease in the quality of life of diabetic population, and a great research incentive.[217]
DFI is defined as the presence of an inflammatory response and tissue damage that can drive the clinical spectrum from superficial cellulitis (mild infection) to chronic osteomyelitis (severe infection), with host–microorganism interaction being crucial in determining progression.[218,219] This interaction is defined by Casadevall and Pirofsk as the “damage response framework model”,[220] and proposes that infection outcomes are dependent on mutual contributions of both the microbe and the host.[220] In comparative terms, infection occurs when invading organisms overwhelm the host’s defenses.[221] In contrast, colonization is defined as the presence of proliferating bacteria without an overt host immunological reaction.[26] The reported critical limit is 105 colony-forming units per gram of tissue,[222] indicating the presence of a ‘critical’ degree of colonization marking the point at which host defenses are no longer able to contain the infection.[223] Nevertheless, preexisting diabetic neuropathy, peripheral vascular disease, impaired leukocyte function[224] and a deteriorated innate immune system make DFI clinical diagnosis difficult while simultaneously worsening its prognosis.[225,226] Under these circumstances, onset of classic signs of infection may not occur,[227] even when there is a high bacterial load.[226] Thus, the invading pathogen may progress and infect with no clinical translation.
Staphylococcus aureus is a major pathogen of human skin. This is also the most common pathogen identified in patients with acute superficial DFU.[23,228,229] This pathogen, in its interaction with the host’s diabetic wound environment, is able to amplify certain glucose-regulated genes that ultimately increase its virulence, indicating that hyperglycemic conditions facilitate pathogen adaptation and survival, ultimately worsening patient prognosis.[230]
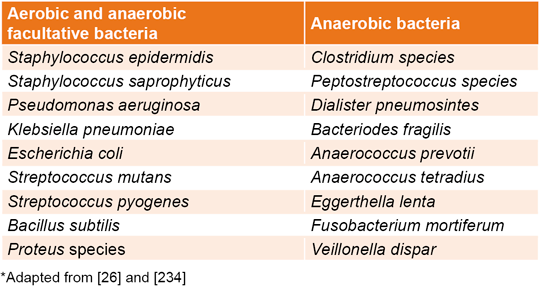
Chronic ulcers usually exhibit polymicrobial infections including a mixture of aerobic/anaerobic and Gram positive/negative bacteria.[227,231] Some of the heterogeneous groups of bacterial species identified in DFU patients have been compiled in Table 1.[26,232–234] Bacterial predominance differs between studies. Nevertheless, recent literature point to Gram-negative bacteria as the predominant group.[23,229,235,236] The reported prevalence included Pseudomonas aeruginosa, Klebsiella pneumoniae, and Escherichia coli.[237,238] Usually, these results are influenced by several factors including infection severity, demographic characteristics, glycemic control, and ongoing or previous antibiotic treatments, as well as bacterial identification method.[26]
Table 1: Bacterial species identified in diabetic foot ulcers
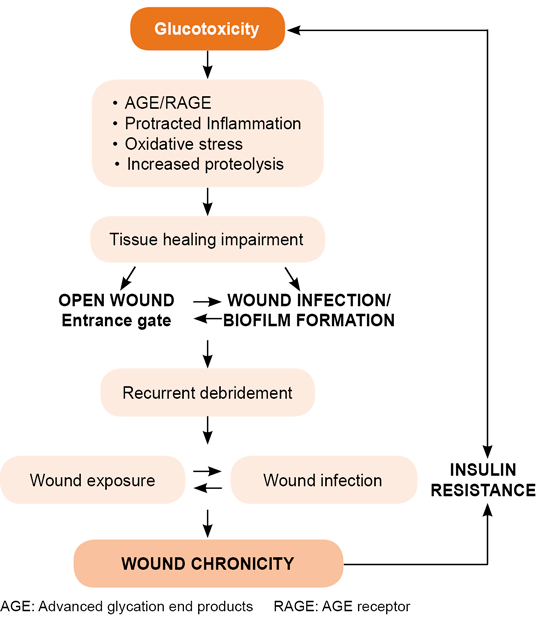
A traditional debate in this field is the relationship between pathogenic potential and microbial bioburden, and how it impacts the host. Here, it is important to highlight the diversity of the microorganism population and its potential interactions, as these may turn into cooperative pathogenic loops that enhance antimicrobial resistance and imprint a particular signature on each individual ulcer.[239] The symbiotic microbial interaction within the ulcer’s ecosystem confers a virulence profile that is far more important than the microorganism concentration in and of itself.[222,240] Long-standing ulcers are more predisposed to infection which, in addition to impairing the healing response, may reduce peripheral insulin sensitivity.[21,241,242] Thus, the longer the wound is maintained, the greater the risk for infection. Another contribution of infected ulcers is their pathogenic effect at the organism level. These ulcers act as pro-inflammatory organs superimposed onto a host, pouring pro-inflammatory reactants, oxygen free radicals and bacterial toxins into central circulation, amplifying tissue injury and general homeostasis (Figure 5).[243]
Finally, it is noteworthy that DFI is frequently associated with existing biofilm in the context of ulcers. Biofilm is a niche for symbiotic microorganism interactions that essentially act as a protector shield for bacterial populations.[244–246] How biofilm-making microorganisms interact with immunocompromised diabetics and their subsequent pathophysiological consequences are relevant research topics.
The term biofilm was coined by the scientific community at the end of the 20th century, which indicates that this is an emerging and expanding research area.[247–250] DFU pathogens can exist as planktonic form (free-living) or as a biofilm (sessile-living).[246] Both phenotypic states may play important roles in impairing healing and causing infection of both acute and chronic wounds.[251] Biofilm acts as a collective entity endowed with superior antimicrobial resistance when compared with its individual constituents. Several in vitro experiments indicate that antibiotic resistance in biofilm bacteria is up to 1000 times higher than in planktonic bacteria.[252]
Figure 5. Wound–infection feedback loop
The ability of a microorganism to build up biofilms is an important virulence factor and an advantageous organizational step. As stated above, biofilm offers a protective environment or physical barrier to biological and antimicrobial substances, facilitating microorganism attachment to surfaces or to each other, ultimately enabling survival and antibiotic resistance.[236,245] ‘Inoffensive’ non-pathogenic bacteria, incapable of promoting chronic wound infection, may symbiotically interlink with pathogenic biofilm and act synergistically to cause a chronic infection.[24] This structured community of microorganisms can be classified as mono- or polymicrobial, encased in extracellular polymeric substances (EPS) or exo-polymeric substances.[253]
The community is a mixture not only of bacterial cells, but also of fungi, viruses, proteins, extracellular DNA and other biogenic factors that increase virulence and reduce treatment success.[246] This and other virulence factors are possible through cell-to-cell communication via quorum sensing (QS).[246,254] QS is a form of cellular communication mediated by small molecules that depend on cell density. The species of bacteria that reaches critical-mass concentration produces large amounts of small signaling molecules that modify gene expression. Indeed, bacterial exchange coordination activities are based on this mechanism, according to population size.[246] QS, together with the exchange of genetic material by bacteria in the biofilm, give rise to different microorganism phenotypes that ultimately affect ulcers as can be seen in anti-microbial treatment results.[23] The fact that bacteria are not motile in the biofilm context and have lower metabolic and proliferative activities than their planktonic counterparts,[255] makes appropriate antibiotic selection difficult. Many antibiotics used for DFI treatment are only effective against actively dividing cells.[256]
Over the last few years, the concept of biofilm in dynamic reciprocity between wound-bed cells and the host has attracted increasing interest. Correspondingly, it has been proposed that biofilms are responsible for over 90% of all chronic wounds. An electron microscopy study assessing wound tissue biopsies suggested that about 60% of chronic wounds have a biofilm compared to 6% of acute wounds.[249] It is likely that at least half of all chronic wounds develop a biofilm.[248,249] This result indicates the contribution of biofilm to impaired wound healing even when molecular mechanisms underlying biofilm-induced chronification remain poorly understood.[257,258]
The various mechanisms by which biofilm obstructs the healing response include failures in granulative tissue formation and the re-epithelialization trajectory.[259] Accordingly, it is likely that these events are consequences of anti-proliferative signals derived from pathogens and a persistent inflammatory environment[257] that aborts fibroblasts and keratinocyte mitogenic, motogenic and secretory functions.[260] In line with this notion, Trøstrup demonstrated that P. aeruginosa induces a state of cellular quiescence reminiscent of premature senescence.[62] P. aeruginosa also secretes a plethora of proteases resulting in collagen, fibrinogen and elastin degradation, inhibition of PMNs and complement systems, and basement membrane degradation.[261,262] Similarly, proteases secreted by S. aureus also degrade collagen and elastin. The ability to degrade surface-associated adhesins enables bacterial phenotype transition from adhesive to invasive.[263] Inhibition of neutrophil phagocytosis and chemotactic activity is also associated with bacterial wound infection.[264] In this steady inflammatory milieu, PMN-derived elastase and other degradative enzymes increase wound tissue damage, expand wound size and perpetuate chronicity.[62] In-depth studies examining P. aeruginosa and host interaction showed that TLR activation is inhibited by pathogen-derived elastase, which allows evasion of host immunosurveillance.[265] P. aeruginosa-derived rhamnolipids inhibit human beta-defensin secretion by challenged keratinocytes, which contributes to pathogen survival and colonization in compromised epithelia.[266] Furthermore, it has been proposed that biofilm and lipopolysaccharide EPS of Gram-negative bacteria inhibit complement activation, further contributing to evasion of the host’s innate defense system.[267,268] Microorganism-derived PAMP, together with platelet-derived factors stimulate the influx of PMNs and other immune cells, spreading the wound’s pro-inflammatory reactants, increasing the level of ECM-degradative proteases, and consequently curtailing the proliferative phase.[112,269,270] Additionally, infiltrated inflammatory cells in response to bacterial invasion via proinflammatory cytokines and AGE/RAGE axis activation produce large amounts of ROS that act as local causal factors for premature cell senescence.[271,272] In other words, the pathogen manages to prevent otherwise normal PAMP-induced innate immunity activation; and graphically speaking, the DFU turns into a battlefield in which the pre-debilitated diabetic host’s immune system is overwhelmed by biofilm-entrenched microorganisms, thus perpetuating wound arrest.[62,273]
Biofilm identification is complex and dependent on more than simple wound cultures obtained and evaluated using traditional microbiological techniques. More sophisticated and expensive techniques such as light and scanning electron microscopy are required to evaluate biofilm in a wound. Therefore, biofilm presence is often overlooked.[274,275] New molecular techniques including DNA micro-arrays, multiplex real-time polymerase chain reaction and functional metagenomics offer a unique opportunity to characterize biofilm microbiome.[218] These technologies facilitate analysis of a microorganism’s resistance potential and virulence factors.[26]
Conclusively, underlying diabetic complications predispose increased risk of developing DFI and other peripheral tissue infections as compared to the risk in healthy populations. Biofilm-forming microorganisms counteract the host’s defenses, prolong DFU inflammation, deteriorate host anabolism and ultimately increase the risk of amputation (Figure 5).[24,258,276]
CONCLUDING REMARKS Wound healing is a complex biological process consisting of precisely-predetermined overlapping phases integrated in a sequential cascade aimed at morpho-functional restoration in a physiological time window. Successful reparative response requires concerted and cooperative integration of both systemic and local signaling networks and driving forces. DM is an archetypal disease in which a variety of both local and systemic factors combine to disturb most healing phases. Thus, DFUs serve as a model for chronic wounds, an often-devastating diabetic complication, and the first cause of lower-limb amputations worldwide.
Underlying the ulcer’s onset and expansion is a complex interplay of pathogenic vicious circles that turn DFUs into a pro-inflammatory, pro-oxidant, pro-apoptogenic and pro-senescence-inducing organ, superimposed onto a host with an already-debilitated immune response. The failure of diabetic patients’ peripheral immunosurveillance, as in the subsequent elicitation of effector mechanisms, is the foremost contributing factor to DFU infections.
Diabetic dysimmunity is likely a major determining factor on patient outcomes. It seems there is still a long way ahead before we achieve a uniform, comprehensive understanding of the actual immune profile of diabetic individuals. Further studies are needed to disentangle critical contradictions and contemporary conceptual paradoxes. Some of the current critical controversies are highlighted in this paper. Of note, however, is the divergent experimental data on whether hyperglycemia reduces pro-inflammatory cytokines and whether this cytokine dampening accounts for immune failure, as compared with other major ongoing questions in DMT2.
We still do not know how to manipulate hyperinflammation or how to reinstate leukocyte physiology once it is disrupted. At the moment, infection remains a dismal complication of these wounds, protracting pre-existing inflammation, dismantling local immune response, amplifying fibroblast and keratinocyte arrest and further disrupting the host’s internal homeostasis. Given that DFUs are recurrently seeded by biofilm, eliminating infection is a challenging task. This review confirms that, despite the efforts to understand DFU pathophysiology, infection pathology and its interaction with the host, DFU prevalence is rising while any reduction in amputation rates remains modest. Therapeutically promising targets include: 1) Identification and pharmacological manipulation of epigenetic drivers in wound-prolonged inflammation and chronification, even in the ideal scenario of a non-infected ulcer; 2) Ablation of the wound cell-senescent drivers, which could contribute to restoring an acute wound-like closure trajectory; and 3) Characterization of actors and pathways underlying hyperglycemia and insulinopenia-induced diabetic immune failure, and subsequent pharmacological interventions to rebuild the immune system. These academic imperatives must go hand-in-hand with diabetic self-care educational programs, as well as systematic foot and neurological examination by qualified specialists.
ACKNOWLEDGMENTS Dr David G. Armstrong acknowledges the support provided by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, USA. Award Number 1R01124789-01A1.
Berlanga-Acosta JA, Guillén-Nieto GE, Rodríguez-Rodríguez N, Mendoza-Mari Y, Bringas-Vega ML, Berlanga-Saez JO, et al. Cellular senescence as the pathogenic hub of diabetes-related wound chronicity. Front Endocrinol (Lausanne). 2020 Sep 16;11:573032. DOI: 3389/fendo.2020.573032
Iraj B, Khorvash F, Ebneshahidi A, Askari G. Prevention of diabetic foot ulcer. Int J Prev Med. 2013 Mar;4(3):373–6.
Rodrigues Costa RH, Anício Cardoso N, Procópio RJ, Navarro TP, Dardik A, de Loiola Cisneros L. Diabetic foot ulcer carries high amputation and mortality rates, particularly in the presence of advanced age, peripheral artery disease and anemia. Diabetes Metabol Syndr. 2017 Dec;11 Suppl 2:S583–S7.
van Netten JJ, Bus SA, Apelqvist J, Lipsky BA, Hinchliffe RJ, Game F, et al. Definitions and criteria for diabetic foot disease. Diabetes Metab Res Rev. 2020 Mar;36 Suppl 1:e3268. DOI: 1002/dmrr.3268
Lin C-W, Armstrong DG, Lin C-H, Liu P-H, Hung S-Y, Lee S-R, et al. Nationwide trends in the epidemiology of diabetic foot complications and lower-extremity amputation over an 8-year period. BMJ Open Diabetes Res Care. 2019 Oct 11;7(1):e000795. DOI: 1136/bmjdrc-2019-000795
Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. N Engl J Med. 2017 Jun 15;376(24):2367–75. DOI: 10.1056/NEJMra1615439
Armstrong DG, Wrobel J, Robbins JM. Guest Editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J. 2007 Dec;4(4):286–7. DOI: 10.1111/j.1742-481X.2007.00392.x
Martínez-De Jesús FR, Ramos-De la Medina A, Remes-Troche JM, Armstrong DG, Wu SC, Lázaro Martínez JL, et al. Efficacy and safety of neutral pH superoxidised solution in severe diabetic foot infections. Int Wound J. 2007 Dec;4(4):353–62. DOI: 10.1111/j.1742-481X.2007.00363.x
Shin JY, Roh S-G, Sharaf B, Lee N-H. Risk of major limb amputation in diabetic foot ulcer and accompanying disease: a meta-analysis. J Plast Reconstr Aesthet Surg. 2017 Dec;70(12):1681–8.
Armstrong DG, Swerdlow MA, Armstrong AA, Conte MS, Padula WV, Bus SA. Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J Foot Ankle Res. 2020 Mar 24;13(1):16.
Berlanga-Acosta J, Schultz GS, López-Mola E, Guillén-Nieto G, García-Siverio M, Herrera-Martínez L. Glucose toxic effects on granulation tissue productive cells: the diabetics’ impaired healing. BioMed Res Int. 2013;2013:256043. DOI: 10.1155/2013/256043
Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005 Nov 12;366(9498):1736–43.
Ramsey SD, Newton K, Blough D, McCulloch DK, Sandhu N, Reiber GE, et al. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care. 1999 Mar;22(3):382–7.
Prompers L, Huijberts M, Apelqvist J, Jude E, Piaggesi A, Bakker K, et al. High prevalence of ischaemia, infection and serious comorbidity in patients with diabetic foot disease in Europe. Baseline results from the Eurodiale study. Diabetologia. 2007 Jan;50(1):18–25.
Namgoong S, Jung S, Han SK, Jeong SH, Dhong ES, Kim WK. Risk factors for major amputation in hospitalised diabetic foot patients. Int Wound J. 2016 Mar;13 (Suppl 1):13–9.
Kim SY, Kim TH, Choi JY, Kwon YJ, Choi DH, Kim KC, et al. Predictors for amputation in patients with diabetic foot wound. Vasc Specialist Int. 2018 Dec;34(4):109–16.
Sen P, Demirdal T, Emir B. Meta‐analysis of risk factors for amputation in diabetic foot infections. Diabetes Metab Res Rev. 2019 Oct;35(7):e3165.
Alkhatieb M, Mortada H, Aljaaly H. Management of a difficult-to-treat diabetic foot wound complicated by osteomyelitis: a case study. Case Rep Surg. 2020 Jun 15;2020:3971581.
Dryden M, Baguneid M, Eckmann C, Corman S, Stephens J, Solem C, et al. Pathophysiology and burden of infection in patients with diabetes mellitus and peripheral vascular disease: focus on skin and soft-tissue infections. Clin Microbiol Infect. 2015 Sep;21 Suppl 2:S27–S32.
Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: a review of pathogenesis. Indian J Endocrinol Metab. 2012 Mar;16 Suppl 1(Suppl 1):S27–36.
Peleg AY, Weerarathna T, McCarthy JS, Davis TM. Common infections in diabetes: pathogenesis, management and relationship to glycaemic control. Diabetes Metab Res Rev. 2007 Jan;23(1):3–13.
Premanath R, Suresh S, Alva PP, Akash S. Biofilm forming abilities of microorganisms associated with diabetic wound infection: a study from a tertiary care hospital. Biomedical Pharmacol J. 2019 Jan 1;12(2):677–82.
Pouget C, Dunyach-Remy C, Pantel A, Schuldiner S, Sotto A, Lavigne JP. Biofilms in diabetic foot ulcers: significance and clinical relevance. Microorganisms. 2020 Oct 14;8(10):1580.
Bjarnsholt T, Buhlin K, Dufrêne YF, Gomelsky M, Moroni A, Ramstedt M, et al. Biofilm formation–what we can learn from recent developments. J Intern Med. 2018 Oct;284(4):322–45.
Jneid J, Lavigne JP, La Scola B, Cassir N. The diabetic foot microbiota: a review. Human Microbiome J. 2017 Dec;5–6:1–6.
Campoccia D, Mirzaei R, Montanaro L, Arciola CR. Hijacking of immune defenses by biofilms: a multifront strategy. Biofouling. 2019 Nov;35(10):1055–74.
Diabetes: an update on the pandemic and potential solutions. In: Ali MK, Siegel KR, Chandrasekar E, Tandon N, Montoya PA, Mbanya JC, et al, editors. Cardiovascular, Respiratory and Related Disorders. Washington, D.C.: The International Bank for Reconstruction and Development; The World Bank; 2017 Nov 17. Chapter 12.
Jenkins TC, Knepper BC, Jason Moore S, Saveli CC, Pawlowski SW, Perlman DM, et al. Comparison of the microbiology and antibiotic treatment among diabetic and nondiabetic patients hospitalized for cellulitis or cutaneous abscess. J Hosp Med. 2014 Dec;9(12):788–94.
Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B. Impaired leucocyte functions in diabetic patients. Diabet Med. 1997 Jan;14(1):29–
Nojima I, Eikawa S, Tomonobu N, Hada Y, Kajitani N, Teshigawara S, et al. Dysfunction of CD8+ PD-1+ T cells in type 2 diabetes caused by the impairment of metabolism-immune axis. Sci Rep. 2020 Sep 10;10(1):14928.
Price CL, Al Hassi HOS, English NR, Blakemore AIF, Stagg AJ, Knight SC. Methylglyoxal modulates immune responses: relevance to diabetes. J Cell Mol Med. 2010 Jun;14(6B):1806–15.
Ozer A, Altuntas CZ, Izgi K, Bicer F, Hultgren SJ, Liu G, et al. Advanced glycation end products facilitate bacterial adherence in urinary tract infection in diabetic mice. Pathog Dis. 2015 Jul;73(5):ftu004.
Creusot RJ, Postigo-Fernández J, Teteloshvili N. Altered function of antigen-presenting cells in type 1 diabetes: a challenge for antigen-specific immunotherapy? Diabetes. 2018 Aug;67(8):1481–94.
Noor S, Khan RU, Ahmad J. Understanding diabetic foot infection and its management. Diabetes Metab Syndr. 2017 Apr–Jun;11(2):149–56.
Zubair M. Prevalence and interrelationships of foot ulcer, risk-factors and antibiotic resistance in foot ulcers in diabetic populations: a systematic review and meta-analysis. World J Diabetes. 2020 Mar 15;11(3):78–83.
Wolcott RD, Rhoads DD. A study of biofilm-based wound management in subjects with critical limb ischaemia. J Wound Care. 2008 Apr;17(4):145–8, 150–2, 154–5.
Wolcott RD, Cox S. More effective cell-based therapy through biofilm suppression. J Wound Care. 2013 Jan;22(1):26–31.
Mohandas A, Anisha BS, Chennazhi KP, Jayakumar R. Chitosan–hyaluronic acid/VEGF loaded fibrin nanoparticles composite sponges for enhancing angiogenesis in wounds. Colloids Surfaces B Biointerfaces. 2015 Mar 1;127:105–13.
Anisha BS, Biswas R, Chennazhi K, Jayakumar R. Chitosan–hyaluronic acid/nano silver composite sponges for drug resistant bacteria infected diabetic wounds. Int J Biol Macromol. 2013 Nov;62:310–20.
Patel S, Srivastava S, Singh MR, Singh D. Mechanistic insight into diabetic wounds: Pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed Pharmacother. 2019 Apr;112:108615.
Sorg H, Tilkorn DJ, Hager S, Hauser J, Mirastschijski U. Skin wound healing: an update on the current knowledge and concepts. Eur Surg Res. 2017;58(1–2):81–94.
Brown A. Phases of the wound healing process. Nurs Times. 2015 Nov 11–17;111(46):12–3.
Enoch S, Leaper DJ. Basic science of wound healing. Surgery (Oxford). 2005 Feb 1;23(2):37–42.
Velnar T, Bailey T, Smrkolj V. The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res. 2009 Sep–Oct;37(5):1528–42.
Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: a cellular perspective. Physiol Rev. 2019 Jan 1;99(1):665–706.
Landén NX, Li D, Ståhle M. Transition from inflammation to proliferation: a critical step during wound healing. Cell Mol Life Sci. 2016 Oct;73(20):3861–85.
Gethin G. Understanding the inflammatory process in wound healing. British J Community Nurs. 2012;17(Suppl 3):S17–S22.
Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007 Jan–Feb;25(1):9–18.
Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic‐Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008 Sep–Oct;16(5):585–601.
Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nature medicine. 2017 Mar 7;23(3):279–87.
Opneja A, Kapoor S, Stavrou EX. Contribution of platelets, the coagulation and fibrinolytic systems to cutaneous wound healing. Thromb Res. 2019 Jul;179:56–63.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015 Jul;21(7):815–9.
Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. 2016 Apr;65(4):1061–71.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004 Mar;303(5663):1532–5.
Strbo N, Yin N, Stojadinovic O. Innate and adaptive immune responses in wound epithelialization. Adv Wound Care (New Rochelle). 2014 Jul;3(7):492–501.
Rodero MP, Khosrotehrani K. Skin wound healing modulation by macrophages. Int J Clin Exp Pathol. 2010 Jul 25;3(7):643–53.
Wilgus TA. Immune cells in the healing skin wound: influential players at each stage of repair. Pharmacol Res. 2008 Aug;58(2):112–6.
Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukocyte Biol. 2013 Jun;93(6):875–81.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013 Jan;229(2):176–85.
Minutti CM, Knipper JA, Allen JE, Zaiss DMW, editors. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol. 2017 Jan;61:3–11.
Trøstrup H, Laulund ASB, Moser C. Insights into host–pathogen interactions in biofilm-infected wounds reveal possibilities for new treatment strategies. Antibiotics (Basel). 2020 Jul 10;9(7):396.
Häkkinen L, Larjava H, Koivisto L. Granulation tissue formation and remodeling. Endodont Topics. 2011 Mar;24(1):94–129.
Alhajj M, Bansal P, Goyal A. Physiology, Granulation Tissue [Internet]. Florida: StatPearls Publishing LLC; 2020 Nov 2 [cited 2021 Jan 25]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK554402/
Guerra A, Belinha J, Jorge RN. Modelling skin wound healing angiogenesis: a review. J Theor Biol. 2018 Dec 14;459:1–17.
Tonnesen MG, Feng X, Clark RA, editors. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000 Dec;5(1):40–6.
Eming SA, Brachvogel B, Odorisio T, Koch M. Regulation of angiogenesis: wound healing as a model. Prog Histochem Cytochem. 2007;42(3):115–70.
Madonna R, Balistreri CR, Geng Y-J, De Caterina R. Diabetic microangiopathy: pathogenetic insights and novel therapeutic approaches. Vasc Pharmacol. 2017 Mar;90:1–7.
Okonkwo UA, DiPietro LA. Diabetes and wound angiogenesis. Int J Molecular Sci. 2017;18(7):1419.
Desmoulière A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005 Jan–Feb;13(1):7–12.
Ibrahim MM, Chen L, Bond JE, Medina MA, Ren L, Kokosis G, et al. Myofibroblasts contribute to but are not necessary for wound contraction. Lab Investig. 2015 Dec;95(12):1429–38.
Ehrlich HP, Hunt TK. Collagen organization critical role in wound contraction. Adv Wound Care (New Rochelle). 2012 Feb;1(1):3–9.
Muñoz M, Vásquez B, del Sol M. Molecular mechanisms in the process of re-epithelization in wound healing and the action of honey in keratinocytes. Int J Morphol. 2020 Dec;38(6):1700–6.
Ylivinkka I, Wickström SA. BETting against wound healing. Nat Chem Biol. 2021 Mar;17(3):233–5.
Liarte S, Bernabé-García A, Nicolás FJ. Role of TGF-in skin chronic wounds: a keratinocyte perspective. Cells. 2020 Jan 28;9(2):306.
Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, et al. Epithelialization in wound healing: a comprehensive review. Adv Wound Care (New Rochelle). 2014 Jul 1;3(7):445–64.
Martin P. Wound healing–aiming for perfect skin regeneration. Science. 1997 Apr 4;276(5309):75–81.
Cañedo-Dorantes L, Cañedo-Ayala M. Skin acute wound healing: a comprehensive review. Int J Inflam. 2019 Jun 2;2019:3706315. DOI: 1155/2019/3706315
Laplante AF, Germain L, Auger FA, Moulin V. Mechanisms of wound re-epithelialization: hints from a tissue‐engineered reconstructed skin to long‐standing questions. FASEB J. 2001 Nov;15(13):2377–89.
Andasari V, Lü D, Swat M, Feng S, Spill F, Chen L, et al. Computational model of wound healing: EGF secreted by fibroblasts promotes delayed re-epithelialization of epithelial keratinocytes. Integr Biol (Cam). 2018 Oct 15;10(10):605–34.
Kirker KR, James GA. In vitro studies evaluating the effects of biofilms on wound‐healing cells: a review. APMIS. 2017 Apr;125(4):344–52.
Rousselle P, Braye F, Dayan G. Re-epithelialization of adult skin wounds: cellular mechanisms and therapeutic strategies. Adv Drug Deliv Rev. 2019 Jun;146:344–65.
Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, et al. High-glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PLoS One. 2014 Sep 22;9(9):e108182.
Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of L‐ Wound Repair Regen. 1998 Mar–Apr;6(2):135–41.
Spravchikov N, Sizyakov G, Gartsbein M, Accili D, Tennenbaum T, Wertheimer E. Glucose effects on skin keratinocytes: implications for diabetes skin complications. Diabetes. 2001 Jul;50(7):1627–35.
Yamaguchi Y, Yoshikawa K. Cutaneous wound healing: an update. J Dermatol. 2001 Oct;28(10):521–34.
Garraud O, Hozzein WN, Badr G. Wound healing: time to look for intelligent, ‘natural’ immunological approaches? BMC Immunol. 2017 Jun 21;18(Suppl 1):23.
Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med. 2006 Jun;23(6):594–608.
Sorg H, Tilkorn DJ, Hager S, Hauser J, Mirastschijski U. Skin wound healing: an update on the current knowledge and concepts. Eur Surg Res. 2017;58(1–2):81–94.
Greenhalgh DG. The role of apoptosis in wound healing. Int J Biochem Cell Biol. 1998 Sep;30(9):1019–30.
Stroncek JD, Reichert WM, Reichert WM, editors. Overview of wound healing in different tissue types. In: Indwelling neural implants: strategies for contending with the in vivo environment. Florida: CRC Press/Taylor & Francis; 2008. Chapter 1. p. 3–41.
Lazarus GS, Cooper DM, Knighton DR, Percoraro RE, Rodeheaver G, Robson MC. Definitions and guidelines for assessment of wounds and evaluation of healing. Wound Repair Regen. 1994 Jul;2(3):165–70.
Stupin VA, Gabitov RB, Sinelnikova TG, Silina EV. Biological mechanisms of chronic wound and diabetic foot healing: the role of collagen. Serbian J Exp Clin Res. 2018 Dec 26;19(4):373–82.
Posnett J, Franks PJ. The burden of chronic wounds in the UK. Nurs Times. 2008 Jan 22–28;104(3):44–5.
Lazaro J, Izzo V, Meaume S, Davies AH, Lobmann R, Uccioli L. Elevated levels of matrix metalloproteinases and chronic wound healing: an updated review of clinical evidence. J Wound Care. 2016 May;25(5):277–87.
Alavi A, Sibbald RG, Mayer D, Goodman L, Botros M, Armstrong DG, et al. Diabetic foot ulcers: Part I. Pathophysiology and prevention. J Am Acad Dermatol. 2014 Jan;70(1):1.e1–18.
Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. 2006 Sep–Oct;14(5):558–65.
Borst SE. The role of TNF-α in insulin resistance. 2004 Mar–Apr;23(2–3):177–82.
Acosta JB, García del Barco D, Cibrian Vera D, Savigne W, López‐Saura P, Guillén Nieto G, et al. The pro‐inflammatory environment in recalcitrant diabetic foot wounds. Int Wound J. 2008 Oct;5(4):530–9.
Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. 1999 Dec;26(3–4):259–65.
Graves DT, Kayal RA. Diabetic complications and dysregulated innate immunity. Front Biosci. 2008 Jan;13:1227–39.
Kawahito S, Kitahata H, Oshita S. Problems associated with glucose toxicity: role of hyperglycemia-induced oxidative stress. World J Gastroenterol. 2009 Sep 7;15(33):4137–42.
Berlanga-Acosta J, Vázquez-Blomquist D, Cibrián D, Mendoza Y, Ochagavía ME, Miranda J, et al. Growth Hormone Releasing Peptide 6 (GHRP6) reduces liver fibrosis in CCl4 chronically intoxicated rats. Biotecnol Aplic. 2012;29(2):60–72.
Berlanga-Acosta J, Mendoza-Marí Y, Fernández-Mayola M, García-Ojalvo A, Valdés- Pérez C, Savigne-Gutiérrez W. Torpid diabetic wound healing: evidence on the role of epigenetic forces. Int J Diabetes Clin Res. 2015;2(1).
Edmonds M, Manu C, Vas P. The current burden of diabetic foot disease. J Clin Orthop Trauma. 2021 Feb 8;17:88–93.
Pastar I, Ojeh N, Glinos GD, Stojadinovic O, Tomic-Canic M. Physiology and pathophysiology of wound healing in diabetes, editors. In: The Diabetic Foot. Contemporary Diabetes. Berlin: Springer; 2018 Sep. p. 109–30.
Rehman K, Akash MS. Mechanisms of inflammatory responses and development of insulin resistance: how are they interlinked? J Biomed Sci. 2016 Dec 3;23(1):87.
Chen L, Chen R, Wang H, Liang F. Mechanisms linking inflammation to insulin Int J Endocrinol. 2015;2015:508409.
Solis-Herruzo JA, Brenner DA, Chojkier M. Tumor necrosis factor alpha inhibits collagen gene transcription and collagen synthesis in cultured human fibroblasts. J Biol 1988 Apr 25;263(12):5841–5.
Unemori E, Hibbs MS, Amento EP. Constitutive expression of a 92-kD gelatinase (type V collagenase) by rheumatoid synovial fibroblasts and its induction in normal human fibroblasts by inflammatory cytokines. J Clin Invest. 1991 Nov;88(5):1656–62.
Turina M, Miller FN, Tucker C, Polk HC Jr. Effects of hyperglycemia, hyperinsulinemia, and hyperosmolarity on neutrophil apoptosis. Surg Infect (Larchmt). 2006 Apr;7(2):111– 21.
McCarty SM, Percival SL. Proteases and delayed wound healing. Adv Wound Care (New Rochelle). 2013 Oct;2(8):438–47.
Lobmann R, Schultz G, Lehnert H. Proteases and the diabetic foot syndrome: mechanisms and therapeutic implications. Diabetes Care. 2005 Feb;28(2):461–71.
Braund R, Hook S, Medlicott NJ. The role of topical growth factors in chronic wounds. Curr Drug Deliv. 2007 Jul;4(3):195–204.
Schönfelder U, Abel M, Wiegand C, Klemm D, Elsner P, Hipler U-C. Influence of selected wound dressings on PMN elastase in chronic wound fluid and their antioxidative potential in vitro. Biomaterials. 2005 Nov;26(33):6664–73.
Dinh T, Pham H. Emerging treatments in diabetic wound care. Wounds. 2002 Jan;14(1):2–10.
Wang L, Zhou X, Yin Y, Mai Y, Zhang X, Wang D. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH oxidase-dependent pathway in diabetic Front Immunol. 2019 Jan 8;9:3076.
Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013 May 7;17(5):695–708.
Krzyszczyk P, Schloss R, Palmer A, Berthiaume F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front Physiol. 2018 May 1;9:419.
Torres-Castro I, Arroyo-Camarena ÚD, Martínez-Reyes CP, Gómez-Arauz AY, Dueñas- Andrade Y, Hernández-Ruiz J, et al. Human monocytes and macrophages undergo M1- type inflammatory polarization in response to high levels of glucose. Immunol Lett. 2016 Aug;176:81–9.
Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS 2010 Mar 4;5(3):e9539.
Moganti K, Li F, Schmuttermaier C, Riemann S, Klüter H, Gratchev A, et al. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte- derived macrophages. Immunobiology. 2017 Oct;222(10):952–9.
Wicks K, Torbica T, Mace KA. Myeloid cell dysfunction and the pathogenesis of the diabetic chronic wound. Semin Immunol. 2014 Aug;26(4):341–53.
Armstrong DG, Gurtner GC. A histologically hostile environment made more hospitable? Nat Rev Endocrinol. 2018 Sep;14(9):511–2.
Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. 1977 Apr;26(4):284–90.
Armstrong DG, Boulton AJM, Bus SA. Diabetic foot ulcers and their recurrence. New Engl J Med. 2017 Jun 15;376(24):2367–75.
Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000 Aug;321(7258):405–12.
Blazer S, Khankin E, Segev Y, Ofir R, Yalon-Hacohen M, Kra-Oz Z, et al. High glucose- induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun. 2002 Aug 9;296(1):93–101.
Blair M. Diabetes Mellitus Review. Urol Nurs. 2016 Jan–Feb;36(1):27–36.
Berlanga-Acosta JA, Guillén-Nieto GE, Rodríguez-Rodríguez N, Mendoza-Mari Y, Bringas-Vega ML, Berlanga-Sáez JO, et al. Cellular senescence as the pathogenic hub of diabetes-related wound chronicity pathways. Front Endocrinol (Lausanne). 2020 Sep 16;11:573032.
Zubair M, Ahmad J. Role of growth factors and cytokines in diabetic foot ulcer healing: a detailed review. Rev Endocrine Metab Disord. 2019 Jun;20(2):207–17.
Berlanga-Acosta J, Valdéz-Pérez C, Savigne-Gutiérrez W, Mendoza-Marí Y, Franco- Pérez N, Vargas-Machiran E, et al. Cellular and molecular insights into the wound healing mechanism in diabetes. Biotecnol Apl. 2010 Oct;27(4):255–61.
Vlassara H, Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep. 2014 Jan;14(1):453.
Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic Korean J Physiol Pharmacol. 2014 Feb;18(1):1–14.
Wang Z, Shi C. Cellular senescence is a promising target for chronic wounds: a comprehensive review. Burns Trauma. 2020 Jun 23;8:tkaa021.
Wilkinson HN, Hardman MJ. Wound healing: cellular mechanisms and pathological Open Biol. 2020 Sep;10(9):200223.
Berlanga-Acosta J, López-Mola E, Garcia-Siverio M, Guillén-Nieto G, López-Saura P, Valdez-Pérez C, et al. Detrimental impact of acute and chronic glucose burden in wound- healing cells: fibroblasts, myofibroblasts and vascular precursor cells. Biotecnol Aplic. 2012;29(4):208–17.
Morasso MI, Tomic‐Canic M. Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell. 2005 Mar;97(3):173–
Terashi H, Izumi K, Deveci M, Rhodes LM, Marcelo CL. High glucose inhibits human epidermal keratinocyte proliferation for cellular studies on diabetes mellitus. Int Wound J. 2005 Dec;2(4):298–304.
Lan CCE, Liu I-H, Fang A-H, Wen C-H, Wu C-S. Hyperglycaemic conditions decrease cultured keratinocyte mobility: implications for impaired wound healing in patients with Br J Dermatol. 2008 Nov;159(5):1103–15.
Abu-Ashour W, Twells LK, Valcour JE, Gamble J-M. Diabetes and the occurrence of infection in primary care: a matched cohort study. BMC Infect Dis. 2018;18(1):1–8.
Bertoni AG, Saydah S, Brancati FL. Diabetes and the risk of infection-related mortality in the US. Diabetes Care. 2001 Jun;24(6):1044–9.
Tsai S, Clemente-Casares X, Revelo XS, Winer S, Winer DA. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? 2015 Jun;64(6):1886–97.
Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis G-A, Vogiatzi G, Papaioannou S, et al. The role of inflammation in diabetes: current concepts and future Eur Cardiol Rev. 2019 Apr;14(1):50–9.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005 May;115(5):1111–9.
Oguntibeju OO. Type 2 diabetes mellitus, oxidative stress and inflammation: examining the links. International journal of physiology, pathophysiology and pharmacology. 2019;11[3]:45.
Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev 2011 Feb;11(2):98–107.
Nielsen TB, Pantapalangkoor P, Yan J, Luna BM, Dekitani K, Bruhn K, et al. Diabetes exacerbates infection via hyperinflammation by signaling through TLR4 and RAGE. 2017 Aug 22;8(4):e00818-17.
Rayfield EJ, Ault MJ, Keusch GT, Brothers MJ, Nechemias C, Smith H. Infection and diabetes: the case for glucose control. Am J Med. 1982 Mar;72(3):439–50.
Knapp S. Diabetes and infection: is there a link?–A mini-review. Gerontology. 2013;59(2):99–104.
Kraine MR, Tisch RM. The role of environmental factors in insulin-dependent diabetes mellitus: an unresolved issue. Environ Health Perspect. 1999 Oct;107 Suppl 5 (Suppl 5):777–81.
Association AD. Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(Suppl 1):S11–S66.
Rubinstein R, Genaro AM, Motta A, Cremaschi G, Wald MR. Impaired immune responses in streptozotocin‐induced type I diabetes in mice. Involvement of high glucose. Clin Exp Immunol. 2008 Nov;154(2):235–46.
Sohlang MN, Majaw S. Altered VDAC-HK association and apoptosis in mouse peripheral blood lymphocytes exposed to diabetic condition: an in vitro and in vivo study. Arch Physiol Biochem. 2021 Jan 12:1–11.
Arya AK, Tripathi R, Kumar S, Tripathi K. Recent advances on the association of apoptosis in chronic non healing diabetic wound. World J Diabetes. 2014 Dec 15;5(6):756–62.
Stafeev IS, Vorotnikov AV, Ratner EI, Menshikov MY, Parfyonova YV. Latent inflammation and insulin resistance in adipose tissue. Int J Endocrinol. 2017;2017:5076732.
Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin- resistant states. Cold Spring Harb Perspect Biol. 2014 Jan 1;6(1):a009191.
Das AK, Kalra S, Tiwaskar M, Bajaj S, Seshadri K, Chowdhury S, et al. Expert group consensus opinion: Role of anti-inflammatory agents in the management of Type-2 diabetes (T2D). J Assoc Physicians India. 2019 Dec;67(12):65–74.
Nwadiugwu MC. Inflammatory activities in type 2 diabetes patients with co-morbid angiopathies and exploring beneficial interventions: a systematic review. Front Public 2021 Jan 25;8:600427.
Ohno Y, Aoki N, Nishimura A. In vitro production of interleukin-1, interleukin-6, and tumor necrosis factor-alpha in insulin-dependent diabetes mellitus. J Clin Endocrinol 1993 Oct;77(4):1072–7.
Mooradian AD, Reed RL, Meredith KE, Scuderi P. Serum levels of tumor necrosis factor and IL-1α and IL-1β in diabetic patients. Diabetes Care. 1991 Jan;14(1):63–5.
Reinhold D, Ansorge S, Schleicher ED. Elevated glucose levels stimulate transforming growth factor-β1 (TGF-β1), suppress interleukin IL-2, IL-6 and IL-10 production and DNA synthesis in peripheral blood mononuclear cells. Horm Metab Res. 1996 Jun;28(6):267–70.
Spindler MP, Ho AM, Tridgell D, McCulloch‐Olson M, Gersuk V, Ni C, et al. Acute hyperglycemia impairs IL‐6 expression in humans. Immun Inflamm Dis. 2016;4(1):91–7.
Akbari M, Hassan-Zadeh V. Hyperglycemia affects the expression of inflammatory genes in peripheral blood mononuclear cells of patients with Type 2 diabetes. Immunol Invest. 2018 Oct;47(7):654–65.
Glund S, Deshmukh A, Long YC, Moller T, Koistinen HA, Caidahl K, et al. Interleukin- 6 directly increases glucose metabolism in resting human skeletal muscle. 2007 Jun;56(6):1630–7.
Wu R, Liu X, Yin J, Wu H, Cai X, Wang N, et al. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism. 2018 Jun;83:18–24.
Feigerlová E, Battaglia-Hsu S-F. IL-6 signaling in diabetic nephropathy: from pathophysiology to therapeutic perspectives. Cytokine Growth Factor Rev. 2017 Oct;37:57–65.
Mesquida M, Drawnel F, Fauser S. The role of inflammation in diabetic eye disease. Semin Immunopathol. 2019 Jul;41(4):427–45.
Guest CB, Park MJ, Johnson DR, Freund GG. The implication of proinflammatory cytokines in type 2 diabetes. Front Biosci. 2008 May 1;13:5187–94.
Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev. 2020;16(5):442–9.
Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition [EPIC]- Potsdam Study. Diabetes. 2003 Mar;52(3):812–7.
Javeed N, Brown MR, Rakshit K, Her T, Ye Z, Lee JH, et al. Pro-inflammatory cytokines disrupt β-cell circadian clocks in diabetes. bioRxiv [Internet]. Yale: BMJ Yale; 2019 Aug 15 [cited 2021 Jan 5]. Available at: https://www.biorxiv.org/content/10.1101/705210v2.full.pdf
Randeria SN, Thomson GJ, Nell TA, Roberts T, Pretorius E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc Diabetol. 2019;18(1):1–15.
Acharya AB, Thakur S, Muddapur MV, Kulkarni RD. Systemic cytokines in type 2 diabetes mellitus and chronic periodontitis. Curr Diabetes Rev. 2018;14(2):182–8.
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008 Sep 29;205(10):2409–17.
Berlanga-Acosta J, Fernández-Mayola M, Mendoza-Marí Y, García-Ojalvo A, Playford RJ, Guillén-Nieto G. Intralesional infiltrations of cell-free filtrates derived from human diabetic tissues delay the healing process and recreate diabetes histopathological changes in healthy rats. Front Clin Diabetes Health. 2021 Mar;2:617741. DOI: 103389/fcdhc.2021.617741
Keating ST, El-Osta A. Glycemic memories and the epigenetic component of diabetic Curr Diab Rep. 2013 Aug;13(4):574–81.
Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014 May;63(5):1748–62.
Akbari M, Hassan-Zadeh V. The inflammatory effect of epigenetic factors and modifications in type 2 diabetes. Inflammopharmacology. 2020 Apr;28(2):345–62.
Hu R, Xia C-Q, Butfiloski E, Clare-Salzler M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin Immunol. 2018 Oct;195:139–48.
Kumar M, Roe K, Nerurkar PV, Orillo B, Thompson KS, Verma S, et al. Reduced immune cell infiltration and increased pro-inflammatory mediators in the brain of Type 2 diabetic mouse model infected with West Nile virus. J Neuroinflammation. 2014 Apr 21;11:80. DOI: 10.1186/1742-2094-11-80.
Martínez N, Ketheesan N, Martens GW, West K, Lien E, Kornfeld H. Defects in early cell recruitment contribute to the increased susceptibility to respiratory Klebsiella pneumoniae infection in diabetic mice. Microbes Infect. 2016 Oct;18(10):649–55.
Nguyen KT, Seth AK, Hong SJ, Geringer MR, Xie P, Leung KP, et al. Deficient cytokine expression and neutrophil oxidative burst contribute to impaired cutaneous wound healing in diabetic, biofilm‐containing chronic wounds. Wound Repair Regen. 2013 Nov– Dec;21(6):833–41.
Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. 2008 Nov;57(11):3090–8.
Chao W-C, Yen C-L, Wu Y-H, Chen S-Y, Hsieh C-Y, Chang T-C, et al. Increased resistin may suppress reactive oxygen species production and inflammasome activation in type 2 diabetic patients with pulmonary tuberculosis infection. Microbes Infect. 2015 Mar;17(3):195–204.
Perner A, Nielsen SE, Rask-Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. 2003 Apr;29(4):642–5.
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen r adicals. Nat Med. 2007 Jun;13(6):688–94.
Osar Z, Samanci T, Demirel GY, Damci T, Ilkova H. Nicotinamide effects oxidative burst activity of neutrophils in patients with poorly controlled type 2 diabetes mellitus. Exp Diabesity Res. 2004 Apr–Jun;5(2):155–62.
Fakhruddin S, Alanazi W, Jackson KE. Diabetes-induced reactive oxygen species: mechanism of their generation and role in renal injury. J Diabetes Res. 2017;2017:8379327.
Sousa A, Lucas M, Ribeiro D, Correia CM, Silva VLM, Silva AMS, et al. Chalcones as modulators of neutrophil oxidative burst under physiological and high glucose J Nat Prod. 2020 Oct 23;83(10):3131–40.
Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019 Jan;20:247–60.
Stegenga ME, van der Crabben SN, Blümer RM, Levi M, Meijers JCM, Serlie MJ, et al. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood. 2008 Jul 1;112(1):82–9.
Hair PS, Echague CG, Rohn RD, Krishna NK, Nyalwidhe JO, Cunnion KM. Hyperglycemic conditions inhibit C3-mediated immunologic control of Staphylococcus J Transl Med. 2012 Mar 5;10:35.
Jafar N, Edriss H, Nugent K. The effect of short-term hyperglycemia on the innate immune system. Am J Med Sci. 2016 Feb;351(2):201–11.
Joshi MB, Lad A, Prasad ASB, Balakrishnan A, Ramachandra L, Satyamoorthy K, et al. High glucose modulates IL‐6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013 Jul 11;587(14):2241–6.
Pavlou S, Lindsay J, Ingram R, Xu H, Chen M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018 Jul 11;19(1):24.
Liu H-F, Zhang H-J, Hu Q-X, Liu X-Y, Wang Z-Q, Fan J-Y, et al. Altered polarization, morphology, and impaired innate immunity germane to resident peritoneal macrophages in mice with long-term type 2 diabetes. J Biomed Biotechnol. 2012;2012:867023.
Ahmed M, de Winther MPJ, Van den Bossche J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology. 2017 Oct;222(10):937–43.
Jin X, Yao T, Zhou Ze, Zhu J, Zhang S, Hu W, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-κB Biomed Res Int. 2015;2015:732450.
Orliaguet L, Dalmas E, Drareni K, Venteclef N, Alzaid F. Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front Endocrinol (Lausanne). 2020 Feb 19;11:62.
Okizaki S-i, Ito Y, Hosono K, Oba K, Ohkubo H, Amano H, et al. Suppressed recruitment of alternatively activated macrophages reduces TGF-β1 and impairs wound healing in streptozotocin-induced diabetic mice. Biomed Pharmacother. 2015 Mar;70:317–25.
Mauriello CT, Hair PS, Rohn RD, Rister NS, Krishna NK, Cunnion KM. Hyperglycemia inhibits complement-mediated immunological control of S. aureus in a rat model of J Diabetes Res. 2014;2014:762051.
Yano H, Kinoshita M, Fujino K, Nakashima M, Yamamoto Y, Miyazaki H, et al. Insulin treatment directly restores neutrophil phagocytosis and bactericidal activity in diabetic mice and thereby improves surgical site Staphylococcus aureus infection. Infect Immun. 2012 Dec;80(12):4409–16.
Otton R, Soriano FG, Verlengia R, Curi R. Diabetes induces apoptosis in lymphocytes. J 2004 Jul;182(1):145–56.
Gleeson LE, Sheedy FJ. Metabolic reprogramming & inflammation: fuelling the host response to pathogens. Semin Immunol. 2016 Oct;28(5):450–68.
Stentz FB, Kitabchi AE. Activated T lymphocytes in type 2 diabetes: implications from in vitro studies. Curr Drug Targets. 2003 Aug;4(6):493–503.
van Niekerk G, Christowitz C, Conradie D, Engelbrecht A-M. Insulin as an immunomodulatory hormone. Cytokine Growth Factor Rev. 2020 Apr;52:34–44.
van Niekerk G, Dalgleish AG, Joubert F, Joubert A, Engelbrecht A-M. The immuno- oncological implications of insulin. Life Sci. 2020 Jan 1;264:118716.
Kaur P, Choudhury D. Insulin promotes wound healing by inactivating NFkβP50/P65 and activating protein and lipid biosynthesis and alternating pro/anti-inflammatory cytokines dynamics. Biomol Concepts. 2019 Feb 22;10(1):11–24.
Zhu L, Fan L, Zhu Y, Wang Y, Bai H, Yang Q, et al. Insulin antagonizes LPS-induced inflammatory responses by activating SR-A1/ERK axis in macrophages. Inflammation. 2019 Apr;42(2):754–62.
Chauhan P, Saha B. Metabolic regulation of infection and inflammation. Cytokine. 2018 Dec;112:1–11.
Ferracini M, Martins JO, Campos MRM, Anger DBC, Jancar S. Impaired phagocytosis by alveolar macrophages from diabetic rats is related to the deficient coupling of LTs to the FcγR signaling cascade. Mol Immunol. 2010 Jul;47(11–12):1974–80.
Tessaro FHG, Ayala TS, Nolasco EL, Bella LM, Martins JO. Insulin influences LPS- induced TNF-α and IL-6 release through distinct pathways in mouse macrophages from different compartments. Cell Physiol Biochem. 2017;42(5):2093–104.
D’alessandra Y, Chiesa M, Vigorelli V, Ricci V, Rurali E, Raucci A, et al. Diabetes induces a transcriptional signature in bone marrow–derived CD34+ hematopoietic stem cells predictive of their progeny dysfunction. Int J Mol Sci. 2021 Jan 31;22(3):1423.
Senneville É, Lipsky BA, Abbas ZG, Aragón‐Sánchez J, Diggle M, Embil JM, et al. Diagnosis of infection in the foot in diabetes: a systematic review. Diabetes Metab Res 2020 Mar;36 Suppl 1:e3281.
Yazdanpanah L, Shahbazian H, Nazari I, Arti HR, Ahmadi F, Mohammadianinejad SE, et al. Incidence and risk factors of diabetic foot ulcer: a population-based diabetic foot cohort (ADFC study)—two-year follow-up study. Int J Endocrinol. 2018 Mar 15;2018:7631659.
Richard J-L, Sotto A, Lavigne J-P. New insights in diabetic foot infection. World J 2011 Feb 15;2(2):24–32.
Jneid J, Cassir N, Schuldiner S, Jourdan N, Sotto A, Lavigne J-P, et al. Exploring the microbiota of diabetic foot infections with culturomics. Front Cell Infect Microbiol. 2018 Aug 14;8:282.
Lavery LA, Armstrong DG, Murdoch DP, Peters EJG, Lipsky BA. Validation of the Infectious Diseases Society of America’s diabetic foot infection classification system. Clin Infect Dis. 2007 Feb 15;44(4):562–5.
Casadevall A, Pirofski L-a. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol. 2003 Oct;1(1):17–24.
Spichler B, Armstrong HD, Lipsky BA. Microbiology of diabetic foot infections: from Louis Pasteur to ‘crime scene investigation’. BMC Medicine. 2015 Jan 7;13(2).
Dow G, Browne A, Sibbald RG. Infection in chronic wounds: controversies in diagnosis and treatment. Ostomy Wound Manage. 1999 Aug;45(8):23–7, 29–40.
Bjarnsholt T, Kirketerp‐Møller K, Jensen PØ, Madsen KG, Phipps R, Krogfelt K, et al. Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen. 2008 Jan–Feb;16(1):2–10.
Edmonds M. Infection in the neuroischemic foot. Int J Lower Extrem Wounds. 2005 Sep;4(3):145–53.
Armstrong DG, Lavery LA, Sariaya M, Ashry H. Leukocytosis is a poor indicator of acute osteomyelitis of the foot in diabetes mellitus. J Foot Ankle Surg. 1996;35(4):280–3.
Gardner SE, Hillis SL, Frantz RA. Clinical signs of infection in diabetic foot ulcers with high microbial load. Biol Res Nurs. 2009 Oct;11(2):119–28.
Ruke M, Savai J. Diabetic foot infection, biofilm new management strategy. Diabetes Res Open Access. 2019 Nov 11;1(1):7–22.
Bader MS. Diabetic foot infection. Am Fam Physician. 2008;78(1):71–9.
Ogba OM, Nsan E, Eyam ES. Aerobic bacteria associated with diabetic foot ulcers and their susceptibility pattern. Biomed Dermatol. 2019 Feb 1;3(1):1–6.
Abdulrazak A, Bitar ZI, Al-Shamali AA, Mobasher LA. Bacteriological study of diabetic foot infections. J Diabetes Complications. 2005 May–Jun;19(3):138–41.
Lipsky BA, Berendt AR, Deery HG, Embil JM, Joseph WS, Karchmer AW, et al. Diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2004 Oct 1;39(7):885–910.
Richard JL, Lavigne JP, Sotto A. Diabetes and foot infection: more than double trouble. Diabetes Metab Res Rev. 2012 Feb;28 Suppl 1:46–53.
Spichler A, Hurwitz BL, Armstrong DG, Lipsky BA. Microbiology of diabetic foot infections: from Louis Pasteur to ‘crime scene investigation’. BMC Med. 2015 Jan 7;13:2.
Rani V, Nithyalakshmi J. A comparative study of Diabetic and Non-diabetic wound infections with special reference to MRSA and ESBL. Int J Curr Microbiol App Sci. 2014;3(12):546–54.
Banu A, Hassan MMN, Rajkumar J, Srinivasa S. Spectrum of bacteria associated with diabetic foot ulcer and biofilm formation: a prospective study. Australas Med J. 2015 Sep 30;8(9):280–5.
Mossabba Ms, Khan DM. Prevalence of diabetic foot ulcer infections associated with Gram negative bacteria with special reference to drug resistant isolates. Int J Biomed Res. 2016;7(11):765–70.
Ishwarya KM, Neelusree P. Bacteriological profile and their antimicrobial susceptibility from diabetic foot infections in a tertiary care centre from Kancheepuram, India. Saudi J Pathol Microbiol. 2019 Feb 28;4(2):134–41.
Oates A, Bowling FL, Boulton AJM, McBain AJ. Molecular and culture-based assessment of the microbial diversity of diabetic chronic foot wounds and contralateral skin sites. J Clin Microbiol. 2012 Jul;50(7):2263–71.
Lipsky BA, Armstrong DG, Citron DM, Tice AD, Morgenstern DE, Abramson MA. Ertapenem versus piperacillin/tazobactam for diabetic foot infections (SIDESTEP): prospective, randomised, controlled, double-blinded, multicentre trial. Lancet. 2005 Nov 12;366(9498):1695–703.
Scott G. The diabetic foot examination: a positive step in the prevention of diabetic foot ulcers and amputation. Osteopathic Fam Physician. 2013 Mar–Apr;5(2):73–8.
Nicolau DP, Stein GE. Therapeutic options for diabetic foot infections: a review with an emphasis on tissue penetration characteristics. J Am Podiatric Med Assoc. 2010 Jan–Feb;100(1):52–63.
Berlanga-Acosta J, Fernández-Montequín J, Valdés-Pérez C, Savigne-Gutiérrez W, Mendoza-Marí Y, García-Ojalvo A, et al. Diabetic foot ulcers and epidermal growth factor: revisiting the local delivery route for a successful outcome. Biomed Res Int. 2017;2017:2923759.
Lavery LA, Bhavan K, Wukich DK. Biofilm and diabetic foot ulcer healing: all hat and no cattle. Ann Transl Med. 2019 Apr;7(7):159.
Vestby LK, Grønseth T, Simm R, Nesse LL. Bacterial biofilm and its role in the pathogenesis of disease. Antibiotics (Basel). 2020 Feb 3;9(2):59.
Clinton A, Carter T. Chronic wound biofilms: pathogenesis and potential therapies. Lab Med. 2015 Fall;46(4):277–84.
Hurlow J, Bowler PG. Clinical experience with wound biofilm and management: a case Ostomy Wound Manage. 2009 Apr;55(4):38–49.
Kirketerp-Møller K, Jensen PØ, Fazli M, Madsen KG, Pedersen J, Moser C, et al. Distribution, organization, and ecology of bacteria in chronic wounds. J Clin 2008 Aug;46(8):2717–22.
James GA, Swogger E, Wolcott R, Pulcini Ed, Secor P, Sestrich J, et al. Biofilms in chronic wounds. Wound Repair Regen. 2008 Jan –Feb;16(1):37–44.
Nwomeh BC, Yager DR, Cohen IK. Physiology of the chronic wound. Clin Plastic 1998 Jul;25(3):341–56.
Attinger C, Wolcott R. Clinically addressing biofilm in chronic wounds. Adv Wound Care (New Rochelle). 2012 Jun;1(3):127–32.
Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet. 2001 Jul 14;358(9276):135–8.
Percival SL, Walker JT, Hunter PR. Microbiological aspects of biofilms and drinking Florida: CRC Press; 2000 Jun 30. 240 p.
Schuster M, Lostroh CP, Ogi T, Greenberg EP. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome J Bacteriol. 2003 Apr;185(7):2066–79.
O’Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Ann Rev Microbiol. 2000;54:49–79.
Bowen G, Richardson N. Biofilm management in chronic wounds and diabetic foot Diabetic Foot J. 2016;19(4):198–204.
Percival SL, McCarty SM, Lipsky B. Biofilms and wounds: an overview of the evidence. Adv Wound Care. 2015 Jul 1;4(7):373–81.
Metcalf DG, Bowler PG. Biofilm delays wound healing: a review of the evidence. Burns 2013 Jun 18;1(1):5–12.
Trøstrup H, Thomsen K, Christophersen LJ, Hougen HP, Bjarnsholt T, Jensen PØ, et al. P seudomonas aeruginosa biofilm aggravates skin inflammatory response in BALB/c mice in a novel chronic wound model. Wound Repair Regen. 2013 Mar–Apr;21(2):292– 9.
Suleman L. Extracellular bacterial proteases in chronic wounds: a potential therapeutic target? Adv Wound Care. 2016 Oct 1;5(10):455–63.
Prasad ASB, Shruptha P, Prabhu V, Srujan C, Nayak UY, Anuradha CKR, et al. Pseudomonas aeruginosa virulence proteins pseudolysin and protease IV impede cutaneous wound healing. Lab Invest. 2020 Dec;100(12):1532–50.
Berne C, Ducret A, Hardy GG, Brun YV. Adhesins involved in attachment to abiotic surfaces by Gram‐negative bacteria. Microbial Spectr. 2015 Aug;3(4):10.
Pietrocola G, Nobile G, Rindi S, Speziale P. Staphylococcus aureus manipulates innate immunity through own and host-expressed proteases. Front Cell Infect Microbiol. 2017 May 5;7:166.
Van Der Plas MJ, Bhongir RK, Kjellström S, Siller H, Kasetty G, Mörgelin M, et al. Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat Commun. 2016 May 16;7:11567.
Dössel J, Meyer‐Hoffert U, Schröder JM, Gerstel U. Pseudomonas aeruginosa‐derived rhamnolipids subvert the host innate immune response through manipulation of the human beta‐defensin‐2 expression. Cell Microbiol. 2012 Sep;14(9):1364–75.
Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, et al. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement‐mediated opsonization. Cell Microbiol. 2012 Jan;14(1):95– 106.
Schiller NL, Joiner KA. Interaction of complement with serum-sensitive and serum- resistant strains of Pseudomonas aeruginosa. Infect Immun. 1986 Dec;54(3):689–94.
Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care (New Rochelle). 2015 Sep 1;4(9):560–82.
Ochoa M, Rahimi R, Ziaie B. Flexible sensors for chronic wound management. IEEE Rev Biomed Eng. 2014;7:73–86.
Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010 Aug;163(2):257–68.
Demidova-Rice TN, Hamblin MR, Herman IM. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 1: normal and chronic wounds: biology, causes, and approaches to care. Adv Skin Wound Care. 2012;25(7):304–14.
Moser C, Pedersen HT, Lerche CJ, Kolpen M, Line L, Thomsen K, et al. Biofilms and host response–helpful or harmful. APMIS. 2017 Apr;125(4):320–38.
Percival SL, Cutting KF. Biofilms: possible strategies for suppression in chronic wounds. Nurs Stand. 2009 Apr 15–21;23(32):64,66,68.
Costerton W, Veeh R, Shirtliff M, Pasmore M, Post C, Ehrlich G. The application of biofilm science to the study and control of chronic bacterial infections. J Clin 2003 Nov;112(10):1466–77.
Percival SL, Bowler P. Biofilms and their potential role in wound healing. Wounds. 2004 Jul;16(7):234–40.
THE AUTHORS Nadia Rodrίguez-Rodrίguez, microbiologist, Genetic Engineering and Biotechnology Center (CIGB), Havana, Cuba. https://orcid.org/0000-0002-7710-185x
David George Armstrong, podiatric surgeon with a master’s degree in tissue repair and wound healing, and a doctorate in medical sciences, Keck School of Medicine, University of Southern California, USA. https://orcid.org/0000-0001-5533-8648
Jorge Berlanga-Acosta (Corresponding author: jorge.berlanga@cigb.edu.cu), veterinarian with a master’s degree in pathology and a doctorate in pharmacology, CIGB, Havana, Cuba. https://orcid.org/0000-0001-9797-1986
Submitted: March 03, 2021 Approved: July 30, 2021 Disclosures: None
Rodríguez-Rodríguez N, Martínez-Jiménez I, García-Ojalvo A, Mendoza-Mari Y, Guillén-Nieto G, Armstrong D, et al. Wound chronicity, impaired immunity and infection in diabetic patients. MEDICC Rev. 2022 Jan;24(1):44–58. DOI:10.37757/MR2021.V23.N3.8
No new Early Online articles at this time. The most recent articles are listed in the Current Issue Table of Contents. Early Online articles are added as soon as they are available, so please check back later.