INTRODUCTION
Dementia is considered the epidemic of the 21st century, and its main risk factor is advanced age. With rapid population aging, the burden of neurodegenerative diseases has increased, including dementia and, particularly, Alzheimer disease (AD), its most common form.[1–3]
AD is a chronic neurodegenerative disease that progressively impairs cognitive function.[3] Its clinical symptoms include memory loss, language disorders, visual, spatial and behavioral changes. Although various symptoms may occur, memory is always affected. Autopsy with neuropathological examination is the gold standard for AD diagnosis. AD neuropathology is characterized by extracellular deposits of amyloid beta peptide (Aβ) in nerve tissue called amyloid or senile plaques, and intracellular deposits of hyperphosphorylated tau protein, known as neurofibrillary tangles.[4]
Diagnosis of probable AD (pAD) is based on criteria adopted by the US National Institute of Neurological and Communicative Disorders and Stroke and the Association of Alzheimer’s Disease and Related Disorders (NICNDS-ADRDA)[5] and DSM-IV.[6] Patients should have a mini mental state examination score (MMSE) of 0–23 (maximum 30)[7] and a clinical dementia rating (CDR) of 1–3 (1 = mild dementia).[8] These criteria have been used for pAD diagnosis for >30 years in clinical studies with 87% sensitivity and 70% specificity.[9] The most documented genetic risk factor for this disease is the ϵ4 allele of apolipoprotein E, Apo E4.[10]
Dementia afflicts 46.8 million people worldwide (approximately 0.5% of the population) and the number is expected to almost double every 20 years, reaching 74.7 million by 2030 and 131.5 million by 2050. There are more than 9.9 million new cases of dementia every year, a new case every 3.2 seconds. The World Alzheimer Report 2015 estimated social and economic costs of dementia at US$818 billion, and predicted these would reach US$1 trillion by 2018.[1]
Cuba is the second oldest country in Latin America, with approximately 19% of its population aged ≥60 years.[11] By 2020, 1 in 4 Cubans will be aged ≥60 years[12] and about half a million will be aged >80 years.[13] This implies that aging-related diseases such as AD will become a major challenge for the public health system. Cuban studies estimate that 1 in 10 people aged ≥65 years develops some sort of dementia, of which AD is the most common.[14–19]
About 150,000 Cubans suffer from AD or related dementia and an increase to 273,000 cases is expected in 2030 and to 440,000, in 2050. Dementia prevalence currently ranges from 6.4% to 10.2% in people aged ≥65 years,[16] for an incidence of 21.8/1000/year.[17,19,20] Annual mortality among people with dementia is 195.5/1000.[20]
Current concepts of AD pathogenesis include involvement of inflammatory and autoimmune components.[21,22] Several studies have reported immunopositivity to neurons in histological brain sections of AD patients, a fact rarely observed in the same brain regions of age-matched nondemented controls.[22,23] Because neuronal antibodies (Abs) are abundant and common in human sera, to the extent that they have been detected in young nondemented individuals,[23] it may well be that these can only contribute to AD pathogenesis if there is dysfunction of the blood–brain barrier, allowing passage into brain tissue.[22–25] A relationship has been demonstrated between AD development and titers of autoantibodies to certain biomolecules.[26–33]
Aβ plaques are generally considered to be associated with neuronal impairment and loss of synapses.[33,34] Antibodies against total brain protein (TBP) have been shown to penetrate the blood-brain barrier and promote intraneuronal Aβ deposition.[23] Several studies have found altered serum levels of anti-TBP in AD patients.[34] These include: anti-Aβ,[30–32] anti-neurotransmitters, anti-S100, antiglial fibrillary acidic protein,[26] antialdolase (anti-ALD),[27] antiganglioside GM1[28] and anti-oxidized low-density lipoproteins.[29] Because results of different studies are inconsistent, it is difficult to establish a relationship between these autoantibody levels, cognitive impairment and AD diagnosis.[35]
It is not clear whether autoantibodies play a pathogenic and causal role or simply occur as a consequence of disease progression. There would be obvious benefits to identification of an antibody or combination of Abs as diagnostic biomarkers for AD.
Current AD biomarker candidates are either expensive, such as magnetic resonance imaging[36] and positron emission tomography,[37] or invasive, such as those requiring lumbar puncture to collect cerebrospinal fluid (CSF).[38] Although considerable progress has been made in demonstrating these biomarkers’ relationships to AD pathophysiology,[39] less costly and invasive biomarkers (blood, serum) for predicting disease progression are urgently needed. This study´s objective was to assess the association of serum levels of Abs against neuronal antigens (TBP, ALD and Aβ) with cognitive performance in Cubans aged ≥65 years, to explore their potential utility as AD biomarkers.
METHODS
Study design and population This was a cross-sectional pilot study of adults aged ≥65 years living in Havana’s Playa Municipality and in Artemisa Province, immediately southwest of Havana. Methods for data collection and neuropsychological assessment have been published previously for the Playa Dementia and Alzheimer Study (EDAP),[15] carried out in September–December 2003 under the National Program for Care of Persons with Disabilities to estimate incidence of AD and other dementias, as well as their risk factors.
Written informed consent was obtained from participants or a relative or caregiver (informant), who could corroborate or provide information requested during assessment. Clinical evaluation was performed by a physician and another team member (a psychologist or disability specialist). A sociodemographic and risk factor questionnaire was administered, neuropsychological assessment conducted, physical and neurological examinations performed, and a relative or caregiver interviewed. Laboratory tests included complete blood count, glycemia, lipid panel and apolipoprotein E genotype.[40]
Data and blood samples for the Playa group came from 74 EDAP participants[15] randomly selected for a study of the association between Apo E4 and cognitive impairment.[41] From September 2012 through May 2014, the Provincial Service for Comprehensive Community Care of Memory Disorders in San Antonio de los Baños (Artemisa Province) used EDAP methods to assess 270 patients, 146 of whom provided blood samples. The Cuban Neuroscience Center (CNEURO) collected blood for genetic and immunological testing in both Playa and Artemisa. In all, 344 patients were examined, only 220 of whom provided blood samples. Lack of reagents made it impossible to test all available samples, so our analysis is based on 143 participants and their samples, 72 from Playa and 71 from Artemisa.
Diagnosis Neurologically and psychiatrically normal subjects, without any cognitive deficit were considered cognitively normal (CN). Mild cognitive impairment (MCI) was diagnosed by Petersen’s criteria:[42]
- memory alteration, corroborated by a caregiver;
- memory alteration documented with tests and specific scales;
- preservation of general cognitive function;
- preservation of activities of daily living; and
- nonfulfillment with DSM-IV dementia criteria[6]
Patients with pAD were diagnosed according to NICNDS-ADRDA[5] and DSM-IV[6] criteria. MMSE scores ranged from 0 to 23 points and CDRs from 1 to 3. In general, these criteria include:
- onset and progression of insidious dementia, which interferes with activities of daily living;
- absence of other systemic or cerebral disease explaining the symptoms;
- clinically established dementia, documented by psychological testing;
- deficits in two or more areas of knowledge;
- progressive memory and other cognitive function impairment, and
- lack of alteration in level of consciousness (patient awake, with no impairment of consciousness, even drowsiness or confusion).
Among 143 participants assessed, 33 were diagnosed as CN; 52, with MCI and 58, with pAD.
Variables Independent variables were age (years); MMSE score; sex; education (years completed); skin color (white, nonwhite); serum levels of anti-TBP, anti-ALD, and anti-Aβ, expressed in optical density units (OD492nm); and serum positivity for the same three Abs.
According to Marcheco-Teruel’s 2011 report, in Cuba individuals perceived as mestizo and black show the greatest proportion of African ancestry; while those perceived as white have the least.[18] Although the relationship between immunological status and ethnicity remains unclear, it has been reported that autoimmune diseases are more common and severe in Afrodescendant individuals.[43] Therefore, in order to determine, in a simple way, the existence of possible interactions between ethnicity and serum Abs levels assessed in the study, the sample was divided by skin color, into white (suggesting a lower proportion of African ancestry) and nonwhite (mestizo and black, suggesting a higher proportion of African ancestry), instead of the usual classification (white, mestizo, and black) used in Cuba’s census.
Obtaining neuronal antigens Brain antigens were extracted from the brains of adult male Wistar rats by homogenization of tissue previously frozen at –70 °C in extraction buffer (2% SDS, 10% glycerol, 5% 2-mercaptoethanol and 0.125 mol/L Tris HCl, pH 6.8). The homogenized tissue was centrifuged at 10,000g for 10 min and the supernatant recovered.[44]
Determination of serum anti-TBP and anti-ALD levels Serum levels of anti-TBP and anti-ALD were assessed by indirect ELISA as described previously.[27,45] The 96-well plates were coated overnight at 4 °C with 100 µL of antigen 1 μg/mL total brain protein extract or 2 μg/mL aldolase C (Sigma Aldrich, USA) diluted in carbonate-bicarbonate coating buffer (Na2CO3/NaHCO3, pH 9.6). After washing the plate with 0.05% H2O-Tween 80 (volume:volume, v:v), it was blocked with 120 μL of 5% skim milk solution (mass:volume) in 1X PBS and incubated for 1 hour at 37 °C. The plate was washed and 100 μL of each serum, diluted 1:300 (v:v) in 0.05% PBS-Tween 80, were added into each well, in triplicate, and incubated for 1 hour at 37 °C. The presence of Abs was detected by a 1/5000 conjugate of anti-human IgG peroxidase (PA1-28647, Thermo Scientific, USA) in 1% skim milk in PBS-Tween and incubated for 1 hour at 37 °C. Finally, the plate was washed with 0.05% H2O-Tween 80 (v:v), and 100 μL of developing solution (0.01% o-phenylenediamine, OPD; 0.03% H2O2, v:v, in substrate buffer) were added and dark-incubated at room temperature for 15 minutes until color development. To stop the reaction, 50 μL of 2.5 mol/L sulfuric acid was added. OD492nm was recorded on a Suma ELISA plate reader (TecnoSuma International, Cuba). Sera were considered positive if OD492nm exceeded the CN group’s mean OD492nm by 1.5 SD.
Determination of serum anti-Aβ levels Serum anti-Aβ levels were assessed by ELISA sandwich assays[46] designed for this purpose. The 96-well plates were coated overnight at 4 °C with 100 μL of human anti-Aβ (1–42) (A3981, Sigma Aldrich, USA), diluted 1/10,000 in carbonate-bicarbonate coating buffer (Na2CO3/NaHCO3, pH 9.6). After washing the plate with 0.05% H2O-Tween 80 (v:v), it was blocked with 120 μL of 5% skim milk solution (m:v) in 1X PBS and incubated for 1 hour at 37 °C. The plate was washed and incubated for 1 hour at 37 °C with 100 μL of rat total brain protein extract with 1 μg/mL concentration. The plate was washed and 100 μL of serum diluted 1/200 in 0.05% PBS-Tween 80 (v:v) were added, in triplicate. It was incubated for 1 hour at 37 °C. The presence of Abs was detected by a conjugate of anti-human IgG with 1/5000 peroxidase (PA1-28647, Thermo Scientific, USA) in 1% skim milk in PBS-Tween, incubated for 1 hour at 37 °C. Finally, the plate was washed with 0.05% H2O-Tween 80 (v:v) and 100 μL of developing solution (0.01% OPD; 0.03% H2O2, v:v, in substrate buffer) were added, and dark-incubated at room temperature for 15 minutes until color development. To stop the reaction, 50 μL of 2.5 mol/L sulfuric acid were added. OD492nm was recorded on a Suma ELISA plate reader (TecnoSuma International, Cuba). Sera were considered positive if OD492nm exceeded the CN group’s mean OD492nm by 1.5 SD.
Analysis A significance threshold of p = 0.05 was selected. Normality of continuous variables was tested using the Kolmogorov-Smirnov test. The means for age and years of schooling were compared using one-way analysis of variance (ANOVA). Comparison of mean autoantibody levels was performed by covariance analysis (ANCOVA), which included age as covariate, taking into account that an increase of autoantibody levels occurs with age, an effect not reported for the other demographic variables studied.[47] For analysis of mean MMSE values by ANCOVA, age and years of education were included, since both variables are known to affect MMSE scores.[48] In cases where a significant effect was detected in ANOVA, the Duncan multiple-rank test was used to identify diagnostic groups that differed from each other. Differences in discrete variable distribution were analyzed using the chi-square test. Receiver operating characteristic curves (ROC) were used to assess predictive power of the parameters obtained in ELISA; the area under the curve (AUC) was used as a measure of discrimination between groups. Hypothesis testing was done according to Hanley and McNeil’s methodology.[49] Diagnostic reference criteria for cognitively normal individuals, mild cognitive impairment and probable AD are provided in the section describing participants. Software packages used were STATISTIC 8.0 and MedCalc 16.2.
Ethics The research protocol was approved by the CNEURO’s Scientific Council and by the Ethics Committee of the National Program for Care of Persons with Disabilities. Participants and/or informants gave informed consent after being provided detailed information about the study’s purpose, procedures and followup, and reassurance that their care would not be affected if they chose not to participate in (or remain in) the study. Data management procedures ensured confidentiality of participant information.
RESULTS
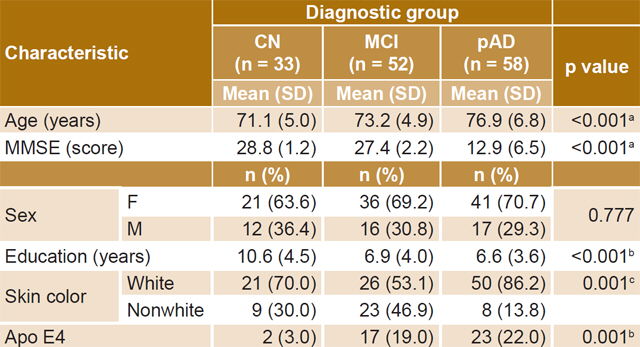
Sample characteristics Table 1 shows mean values for continuous variables and the distribution of discrete variables. Also included is the percentage of individuals in each group with Apo E4. Normality of distribution was confirmed for continuous variables. The Kolmogorov-Smirnov test obtained nonsignificant p values for age (0.257), education (0.074), and serum anti-ALD (0.102), anti-Aβ (0.053) and anti-TBP (0.66).
No statistically significant differences were found between mean ages of the CN and MCI groups (CN 71.0, SD 4.9; MCI 73.2, SD 4.9; Duncan multiple-rank test p = 0.077). However, there were significant differences between the pAD group’s mean age and those of the CN and the MCI group (76.9, SD 6.8; ANOVA p<0.001).
There were no significant differences between diagnostic groups in percentages of men and women (X2 = 0.05, df = 2, p = 0.777). Mean educational level in the sample was 7.65 years, SD 4.21; it was significantly higher in the CN group than in the MCI and pAD groups (CN: 10.6, SD 4.5; MCI: 6.9, SD 4.0; pAD: 6.6, SD 3.6; ANOVA, p<0.001). The latter two did not show significant differences with respect to age (Duncan multiple-rank test, p = 0.743).
Although white skin color predominated in all groups, the proportions of white and nonwhite individuals were significantly different among the three diagnostic groups (X2 = 14.13, df = 2, p<0.001). A higher proportion of individuals with white skin color was observed in the pAD and CN groups, especially in the former. Meanwhile, in the MCI group, white skin color was slightly more common than nonwhite.
Table 1: Sample characteristics by diagnostic group
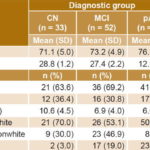
a pAD vs. CN and MCI b CN vs. MCI and pAD c differences among the three diagnostic groups Apo E4: apolipoprotein E4 CN: cognitively normal MCI: mild cognitive impairment MMSE: mini mental state examination pAD: probable Alzheimer disease
Patients with pAD had the lowest MMSE scores (CN: 28.8, SD 1.2; MCI: 27.4, SD 2.2; pAD: 12.9, SD 6.5; ANOVA, p<0.001). Finally, there was a significantly higher frequency of Apo E4 in both the MCI and the pAD groups (X2 = 14.44, df = 2, p = 0.001) compared to CN.
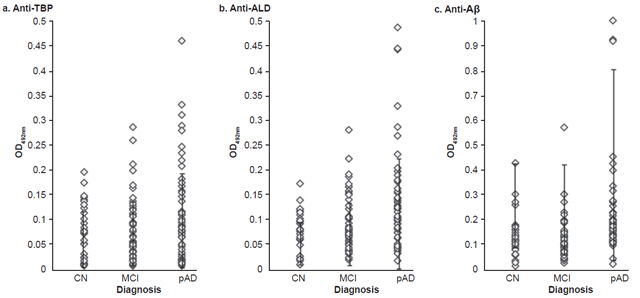
Serum anti-TBP, anti-Aβ and anti-ALD levels Age was consi-dered a covariate in comparison of serum antibody levels. Serum anti-TBP levels did not differ significantly among the three diagnostic groups (mean, SD: CN 0.061, 0.055; MCI 0.071, 0.063; pAD 0.090, 0.085; ANCOVA, p = 0.321). There were no significant differences in serum levels of anti-ALD (mean, SD: CN 0.077, 0.042; MCI 0.095, 0.060; pAD 0.129, 0.120; ANCOVA, p = 0.085). However, serum levels of anti-Aβ were different (mean, SD: CN 0.121, 0.083; MCI 0.107, 0.086; pAD 0.207, 0.205; ANCOVA, p = 0.004) among the three diagnostic groups. Mean OD492nm obtained for anti-Aβ was significantly higher in the pAD group than in the CN (p = 0.007) and MCI (p = 0.002) groups, while significant differences were not found between the CN and MCI groups (p = 0.65) (Figure 1).
The percentage of patients positive for anti-TBP (OD492nm ≥0.143) in the MCI (11.5%) and pAD (24.1%) groups were significantly higher than those in the CN group (6.1%) (X2 = 6.21, df = 2, p = 0.044) (Figure 1a). There were no significant differences in the percentage of individuals positive for anti-ALD (OD492nm ≥0.139) among the three groups (pAD 24.1%, MCI 23.1%, CN 6.1%, X2 = 4.99, df = 2, p = 0.082) (Figure 1b). The percentage of anti-Aβ (19%) in pAD-group patients (OD492nm > 0.245) was significantly higher than those in the MCI (4%) and CN (9.1%) groups, (X2 = 6.19, df = 2, p = 0.045) (Figure 1c).
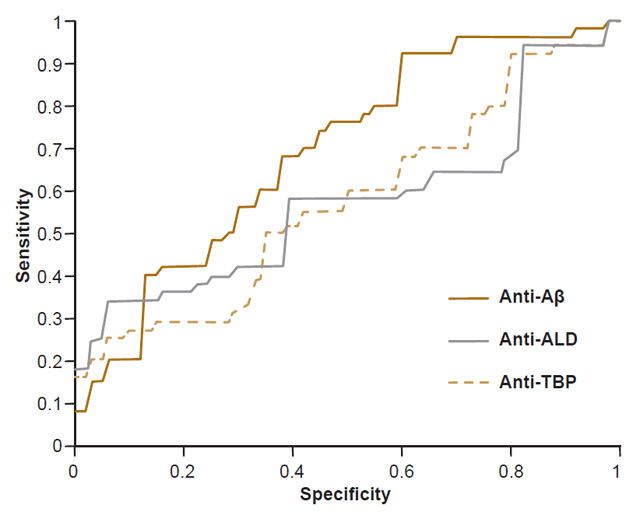
Comparison of predictive power For each diagnostic method, the respective ROCs were constructed and sensitivity and specificity values determined, choosing a cutoff point to maximize the Youden index (sensitivity + specificity – 1).[50]
The predictive power of anti-Aβ and anti-ALD values obtained in the assays was assessed by analyzing their ROC curves (Figure 2). Determination of serum levels of anti-Aβ showed sensitivity of 67% and specificity of 63% (AUC, 0.70; 95% CI 0.59 – 0.79). Subsequent analysis compared diagnostic accuracy with preset cutoff points of 80% for sensitivity and specificity. For 80% sensitivity, specificity was 45%, and for 80% specificity, sensitivity was 41%. The assay used to determine serum levels of anti-ALD had 55% sensitivity and 60% specificity (AUC 0.57, 95% CI 0.46 – 0.67) with sensitivity of 34% and specificity of 21%, with their counterpart set at 80%. Finally, the assay used for to determine serum anti-TBP levels had 48% sensitivity and 63% specificity (AUC 0.58, 95% CI 0.47 – 0.68), with sensitivity of 27% and specificity of 27% with their counterpart set at 80%.
Figure 1: Serum antibody levels by diagnostic group

Aβ: amyloid beta ALD: aldolase CN: cognitively normal MCI: mild cognitive impairment OD492nm: optical density units at 492 nm pAD: probable Alzheimer disease TBP: total brain protein
Figure 2: ELISA ROC curves for serum levels of anti-Aβ, anti-ALD and anti-TBP
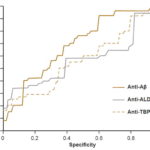
Aβ: amyloid beta ALD: aldolase TBP: total brain protein
DISCUSSION
It is important to have biomarkers as diagnostic resources for early and presymptomatic identification of patients with AD, to facilitate decisions on individualized treatment and followup, as well as to improve development of drugs for this disorder. The diagnostic process begins with tests that have high sensitivity but low specificity (and low cost) and continues with other more specific ones useful for longitudinal quantification of the benefit of a given treatment. Biomarkers for AD should be directly correlated with the disease’s pathophysiology. They may be compounds obtained from body fluids or tissues (such as the tests described in cerebrospinal fluid) or brain images. This was a cross-sectional pilot study that enabled us to propose evaluation of a biomarker associated to formation of Aβ plaques. Our literature review found no such studies in Latin America or the Caribbean.
Serum levels of anti-TBP, anti-ALD and anti-Aβ Similarly to Levin’s findings,[23] we detected no significant differences between anti-TBP levels in CN individuals and those with MCI or pAD. However, the percentage of positive individuals in the pAD group is significantly higher than in the CN and MCI groups. This suggests a possible relationship of anti-TBP with AD and is compatible with the mechanism proposed by Nagele[25] to explain the possible contribution of serum anti-TBP to AD pathogenesis: binding anti-Aβ to the neuronal surface activates receptor-mediated endocytosis, facilitating internalization and chronic accumulation of Aβ, which eventually causes neuronal death and release of amyloid aggregates into the extracellular space.
On the other hand, Mor identified aldolase as the main autoantigen in patients with AD,[27] a result inconsistent with the lack of significant differences between cognitively normal patients and the others in mean anti-ALD levels and percentage of anti-ALD positivity. It has been argued that anti-ALD could be derived from autoimmunization due to ongoing neuronal damage during AD development. The inhibitory activity of AD patients’ sera on aldolase enzyme activity observed by Mor[27] led him to theorize that anti-ALD could enter living neurons and affect ATP production. While Douglas proved that internalization of intracellular anti-TBP in susceptible neurons is possible via Fcγ receptors, and described its harmful effects,[51] Mor’s work did not confirm the ability of anti-ALD to enter living neurons, nor did it assess the effect of anti-ALD sera or Abs on energy metabolism in neuron cultures.[27]
Since Mor’s is the only work that describes a relationship between AD and serum levels of anti-ALD,[27] there is limited scope for assessing the reproducibility of our results. However, Dale found increased levels of anti-ALD in patients with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS), and there were indications that these Abs could even become pathogenic, causing cell apoptosis.[45] The absence of homology between human neuronal aldolase and the aldolase of Streptococcus pyogenes rules out, the possibility that anti-ALD were caused by molecular mimicry in that study.
Given this background, it is likely that anti-ALD are the result of autoimmunization resulting from neuronal damage, and not primary immunological components in the pathogenesis of PANDAS and AD. Even so, anti-ALD could be useful as biomarkers of neuronal damage, to monitor neurological impairment associated with different diseases.
Natural anti-Aβ have generated great interest as AD biomarkers because they are detected in both serum and CSF.[25,30–32] Some authors attribute a protective role to them, because of their effects on Aβ elimination in transgenic mice,[52] and their neuroprotective activity.[22] Others consider them risk factors, due to the correlation detected between anti-Aβ plasma levels and Aβ deposits in the brain.[25] There is great heterogeneity in reports on serum levels of anti-Aβ in AD patients and there is still no consensus on the nature of their relationship to the disease.[53–57]
Discrepancies in these results could be a product of different experimental designs and possible underestimation of serum levels of anti-Aβ, because those that form immunocomplexes in blood are undetectable. Gustaw-Rothenberg reported a higher level of anti-Aβ in serum of AD patients by performing acidic dissociation of immunocomplexes,[53] and Storace detected elevated serum levels of anti-Aβ in patients with MCI who progressed to AD.[54] However, even results of studies using acid dissociation are heterogeneous; e.g., Klaver’s replication failed to find such differences.[55]
Maftei examined serum from healthy adults aged 18–89 years and found that immunocomplexes were more abundant than free anti-Aβ and their levels significantly higher in patients with AD.[56] This was consistent with Gustaw-Rothenberg’s findings using acid dissociation[53] and Mruthinti’s using immunopurification of serum anti-Aβ),[57] as well as with our results using sandwich ELISA. Our finding that mean OD492nm for anti-Aβ and percentage of anti-Aβ positivity were significantly higher in the pAD group than in the CN group is consistent with Nath’s conclusion about the ability of anti-Aβ to magnify Aβ’s neurotoxicity.[32]
Unlike previous studies,[53–57] ours used natural murine Aβ as substitute for synthetic Aβ. According to Dale, autoantibodies developed in patients equally recognize human or murine neuroantigens,[45] making murine Aβ an acceptable alternative to synthetic Aβ or natural human Aβ.
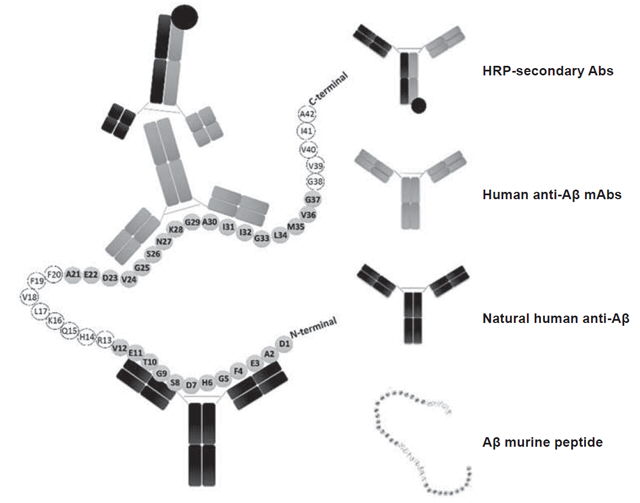
In the sandwich ELISA design we employed, natural murine Aβ is captured by a commercial anti-Aβ (Sigma, A3981, USA) previously immobilized on ELISA plate wells (Figure 3). This commercial anti-Aβ recognizes amino acid residues between positions 1 and 12 near the N-terminal end of Aβ, whereas natural anti-Aβ in human serum recognize amino acid residues between positions 21 and 37 near the C-terminal region.[52,56] In the sandwich ELISA we used, differential recognition (without interference) of Aβ by these two Abs was used to determine serum levels of anti-Aβ. This assay, unlike the ELISA developed by Maftei,[56] allows measurement of serum levels of free anti-Aβ and not those forming immunocomplexes.
Figure 3: Sandwich ELISA design for detection of serum anti-Aβ
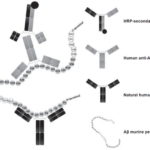
Abs: antibodies Aβ: beta amyloid HRP: horseradish peroxidase mAbs: monoclonal antibodies Note: Amino acids included in regions recognized by human and commercial anti-Aβ are shown in a darker color with their corresponding antibodies.
In both ROC analysis and comparison of mean serum antibody levels, testing for anti-Aβ was the only one with acceptable diagnostic properties for distinguishing between individuals with pAD and those who were cognitively normal. Specificity and AUC values we obtained using ELISA for determination of anti-Aβ are similar to those observed by Maftei,[56] suggesting the usefulness of both methods for molecular diagnosis of AD.
Several studies report alterations of serum levels of anti-TBP in AD patients,[30–33] Such alterations could cause or predispose to development of AD;[25] or they could be due to autoimmunization resulting from cell damage induced by cytotoxicity of Aβ plaques;[32] or they could be part of a protective immune response, by helping remove Aβ plaques.[24] If they are to be used as biomarkers, it will be essential to clarify the precise nature of their relationship to AD.
The generally low percentages of individuals positive for anti-TBP, anti-Aβ and anti-ALD and the data dispersion found in this study could also have different explanations. They could be due to individuals in the AD group with low serum antibody levels in fact having another type of dementia; to loss of serum reactivity in some patients because of epitope or determinant dispersion;[58,59] and/or to presence of Aβ-anti-Aβ immunocomplexes[53] undetectable by the ELISA design we used. It is also worth pointing out that these same factors would affect ROC curve results. We propose validating the method with molecular imaging studies, which provide greater certainty in AD diagnosis.
The ability of this ELISA to detect differences in serum levels of anti-Aβ among the three diagnostic groups suggests its usefulness for an alternative or complementary approach to previously reported direct ELISAs.[22–25] It does not require additional sample preparation steps, such as acid dissociation or immunopurification, and can provide information about anti-Aβ’s affinity and the behavior of free Abs at different disease stages.
Study limitations and future directions Pilot studies such as this represent a fundamental phase in the research process, examining the feasibility of new methods intended for use in a larger study. It should be noted that pilot studies are not hypothesis testing, nor do they assess safety, efficacy or effectiveness. Because sample sizes are limited, they cannot provide reliable estimates of effect size for power calculations for later studies. What they can do is provide evidence of feasibility and identify changes needed in the experimental design.[60]
This study was limited by its small sample size and by differences in ethnic composition (for which distribution of skin color was considered a surrogate), so that participants may not be representative of the general population. It was also limited by its cross-sectional design. A confirmatory study with larger sample sizes and longitudinal followup is required to assess the effects of differing ethnic mix and determine the predictive value of anti-Aβ levels for diagnosis and early detection of cognitive impairment and AD. It will also be necessary to include molecular imaging studies, in addition to neuropsychological diagnosis, to validate the diagnostic power of the method used.
CONCLUSIONS
Our results support a relationship between anti-Aβ and AD pathology. The diagnostic performance and predictive value of serum anti-Aβ suggest they could be useful as biomarkers in early diagnosis of AD. A larger and more detailed study would clarify the relationships between AD and such Abs, as well as their optimal use. Validation and improvement of this noninvasive method would have a substantial clinical impact, since it would permit population monitoring to identify vulnerable groups and improve prevention efforts.