A NEW PROBLEM WITH OLD ROOTS
Since 2004, the term “biosimilar” has been used in scientific and health policy publications to refer to a biological drug produced by biotechnology methods with a pharmacological structure and effects similar to those of a product already on the market, thus usable for the same indications as the original product.[1] There is a complex, politically charged debate around use of biosimilars because these similar medications have new characteristics; they do not fit within traditional frameworks or previous regulatory contexts.
This issue did not exist before the turn of the 21st century. The few biotech drugs that did exist were protected by patents, and technology required to obtain them was controlled by a few companies and countries. This panorama changed with the expansion of biotech products in the pharmacopoeia and the expiration of patents protecting those first products, which still generate millions of dollars in sales.[2] Very few countries can hope to meet the needs of their patients if they are obliged to use expensive, imported products for long-term use. The costs are unsustainable.
One possible answer is to allow onto the market copies of an original medication manufactured by new producers, as happened 30 years ago with generics. This could increase access to products and reduce prices. The complexity of the system for manufacturing biological products makes it almost impossible to make an exact copy of another molecule obtained from a different industrial process. Furthermore, biological products are molecularly quite complex. Even with today’s modern analytical technology, it is difficult to characterize them fully and understand the impact of structural differences between the original molecule and a potential biosimilar on product efficacy and safety.[3]
The current debate is a power struggle. Companies with biotech production capacity and countries with inadequate availability of products lobby for their biosimilars to be registered and sold, while companies manufacturing original biopharmaceuticals, which to date have benefited from a patent-protected monopoly, lobby for stricter regulations to preserve the status quo of few producers and high prices. Outside Europe, the USA and Japan, there are only a few companies in India, China, Brazil, South Korea and Cuba able to produce these types of biosimilars.
Biotechnology companies from low- and middle-income countries cannot implement their own independent strategies to face this challenge. National and regional strategies must be implemented overseen by ministries of health and supported by civil society to guarantee the broadest coverage possible in the best public interest. To design such strategies and gain consensus, we must understand the roots and subtleties of the problem, separate core issues from situational or peripheral ones, distinguish between technical and socioeconomic problems, and establish a space for potential alternatives.
This article aims to stimulate a debate on these strategies.
CONSIDERATIONS FOR DEBATE
1. Biotech products increasingly occupy more space in the pharmacopeia.
Until the 1970s, biological products consisted mainly of vaccines and blood products. They occupied less than 10% of the pharmaceutical market.[2]
The rise of cloning and gene expression technology propelled the biotechnology revolution of the 1980s. Several companies began to develop increasingly complex molecules produced by biosynthesis of genetically modified organisms. The biotechnology segment of the pharmaceutical industry took off with approval of Genentech’s recombinant human insulin in 1982.[4] The first therapeutic monoclonal antibody approved by the US Food and Drug Administration (FDA) was muromonab, used to treat transplant rejection, in 1986. The first recombinant monoclonal antibody for cancer treatment was registered in 1997. Today, 208 biological products are registered, occupying about 25% of the market, expected to reach 50% over the next several years.[2]
In many countries, drug patents were not considered valid until 1994. It was thought that the drug market should be free, and the entry of new competitors favored lower prices and more access. The General Agreement on Trade and Tariffs negotiations ended that year, leading to the creation of the World Trade Organization in 1995. Pressure from powerful multinational pharmaceutical companies imposed universal patent protection for medications.[5]
In the early 21st century, the most important drug patents began to expire after their 20-year terms, providing a legal space for entry of biosimilars.
2 .The current regulatory controversy dates to the introduction of chemical generics in 1984.
The US Pharmacopeia, the first compendium of medications, was established in 1820. At the end of the 19th century, the main concern regarding medication quality was stability.[6] The regulatory functions of the FDA began with the Pure Food and Drug Act (PFDA) of 1906, which prevented sale of adulterated and mislabeled food and drugs.[6] In 1937, there were 105 deaths associated with taking medication that used diethylene glycol as a solvent, and additional safety requirements were added in 1938, marking the beginning of FDA regulation of nonbiological medication manufacture.[6]
In the first half of the 20th century, medications were small molecules created from chemical synthesis. Due to increased knowledge of synthesis mechanisms, more companies began manufacturing generics (medications sold under the name of its main active ingredient with the same composition, pharmaceutical form and bioavailability) after patents expired. In the mid 1950s, increased analytical capacity made generic drug evaluation possible through bioavailability measurements.[7,8] Drug safety was the only criteria for their approval. For a generic medication to be approved, the FDA required scientific publications demonstrating their safety.[6]
In 1961, it was determined that thalidomide was the cause of phocomelia, a congenital malformation, spurring the introduction in 1962 of the Kefauver–Harris Amendment to the PFDA; it added requirements for efficacy, informed consent of study subjects, adverse event reporting, and good manufacturing practices.[6] These new requirements increased time and costs of registration, which, in turn, reduced the number of generics submitted to regulatory agencies for evaluation. In 1963, only 15 applications were submitted to FDA for generics, despite 150 drug patents having expired that year.[6]
Pioneering studies of bioavailability and bioequivalence were conducted in the USA in the 1970s. Based on these, in 1977 FDA incorporated the two concepts in the regulatory process.[7,8] In 1984, the Hatch–Waxman Act (Drug Price Competition and Patent Term Restoration Act) was introduced to stimulate price competition among medications after their patents expired. The Act eased requirements for submitting information and established a simpler procedure for approving generic copies of original medications.[9,10] It established that a medication containing the same active ingredients as the original (the other ingredients could vary) with identical strength, dose, route of administration and indications, with the same identity, safety and purity requirements, demonstrating bioequivalence, was a generic product and could be marketed as such after its manufacturing facilities passed inspection, without the need for additional preclinical or clinical studies.[10]
Small molecules are relatively easy to obtain using reverse engineering techniques. When a patent expires, prices drop to around marginal costs. Today, generics represent 86% of all prescriptions in the USA, with sales amounting to 1.2 quintillion dollars.[11] The Hatch–Waxman Act ushered in a new era in the pharmaceutical industry.[12,13] Entry of generics was compensated by a protective mechanism for the original producer, five years of data exclusivity, activated on FDA approval of a drug that is a new chemical entity. During this period, the FDA cannot approve a generic version.[9]
3. The regulatory environment for biosimilars continues to be confusing and evolving.
The first biotech drug patents to expire were for relatively small molecules, such as insulin, filgrastim and erythropoietin, with molecular weights of 5808, 18,800 and 30,400 mass units, respectively. Monoclonal antibodies are larger molecules (molecular weight of some 150,000 mass units) with greater molecular heterogeneity.[11]
In 2009, the US Biologics Price Competition and Innovation Act established 12.5-year data exclusivity after the sales period began, giving additional protection to the developer when the time remaining on the patent is shorter than the data exclusivity period.[14] A provision was established to ensure that biological products were not treated as “generics.” Clinical trials had to be repeated, comparing them with the original product. This requirement, of course, drives up development costs, and hence, prices.
Although many pharmaceutical companies can imitate original medications based on chemical products, far fewer have the capacity and financial capital to copy biological drugs. Biotech drugs are seeing the highest sales growth in the pharmaceutical sector, more than doubling between 2006 and 2016, from $93 to $200 trillion, and constituting approximately 16% of total medication sales in the latter year.[11] It has been predicted that by 2020, biologics will make up half of all medical prescriptions worldwide.[3]
Unlike synthetic chemical products, the molecular identity of which can be established, biological products obtained through recombinant DNA techniques are intrinsically variable and there is no clear consensus on how molecularly similar a product must be to ensure clinical equivalence. We also lack knowledge of the relationships between manufacturing processes and their resulting products, and between molecular structures and their biological functions.
Another controversial topic is that of statistical methods for evaluating biosimilars. One approach uses Bayesian statistics[15] to explicitly demonstrate the influence of prior evidence on data analysis, via an a priori probability function. This method can be adjusted to the case of biosimilars: when evaluating a product, there is a large amount of prior evidence derived from experience using the original product. Standard, or “frequentist,” statistical methods, however, are created to record evidence from trials one by one, without explicitly including prior evidence in analytical algorithms.
4. There is a trend towards restricting entry of biosimilars into the market.
Large pharmaceutical companies have responded to the expiration of their first-line biological product patents with pressure for increasingly demanding regulations, which make patents a barrier to entry of new competitors. That strategy has been successful for corporate interests, but not for the needs of health systems.
In 2005 a guideline for biosimilars product appeared in the European Medicines Agency (EMA);[16] guidelines on clinical and nonclinical quality followed in 2006, and later, various specific product guidelines. The Pharmaceuticals and Medical Devices Agency of Japan published its Guideline for the Quality, Safety, and Efficacy Assurance of Follow-on Biologics in 2009. That same year, the USA introduced the Biologics Price Competition and Innovation Act and in April 2015, one month after approval of the first biosimilar, three guidelines on biosimilars were published. In 2017 the interchangeability guide was published.[17]
Fully 20 years after patents expired, only 5 biosimilars are registered in the USA (contained in a total of 7 products). In Europe, 14 biosimilars (contained in a total of 41 products) have been registered since 2006, and in Japan, 7 approvals have been filed for 5 biosimilars since 2009.
The concepts of comparability (evaluation of an original biologic after a manufacturing change) and biosimilarity (evaluation of a new biosimilar against the original product) are related scientific and regulatory concepts.[3] It makes no sense to forgo comparative clinical studies after a change in manufacturing in the original company, while they are required when the process is handled by another company.
A biosimilar product may be considered interchangeable with its reference product if it meets two conditions: 1) the biosimilar has the same clinical effect as the reference medication for all patients and 2) in the event these products are taken more than once (as is the case with all biologics), substituting it for the reference product entails no additional risks to safety and efficacy compared to exclusive use of the reference product.[17] FDA regulations establish that all interchangeability tests should be preceded by a comparability study of sufficient sample size. This implies that, after costly comparability studies between the original molecule and the potential biosimilar, yet another trial involving many patients would be needed to evaluate interchangeability.
However, to reduce prices, the regulatory stipulations for interchangeability of biosimilars should closely resemble those of the Hatch–Waxman Act’s for establishing a generic medication, so that pharmacists could automatically substitute a biosimilar for the original drug, as occurs with generics.
An in-depth chemical and physical characterization of molecular structure based on an understanding of the relationship between the medication’s structure and its biological function, together with a safety study, is much more sensitive for detecting changes, using Bayesian methods (Figure 1), than clinical efficacy trials. If the characterization is satisfactory, pharmacokinetic, pharmacodynamic and immunogenicity tests would be conducted. The main variables used would be pharmacokinetic parameters, which are more sensitive and shorten the duration of the studies with patients to evaluate the impact of a medication and its interchangeability. This process should be sufficient for approval of a biosimilar product for the indications for which the original product was approved, as long as there is a commitment to carry out extensive postregistration pharmacovigilance studies.
5. Small changes occur to products’ molecular structures with each new batch, and with each transfer to a new manufacturing facility, without requiring new clinical trials.
Figure 1: Proposed evidence strategy for approval of biosimilars
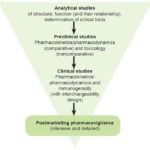
Sandoz and Avalere Health have argued that analytical techniques have progressed to the point that biosimilar manufacturers should provide similarity tests, just as producers of the original molecule test equivalence by analyzing the consequences of changes in production.[18]
Molecular variability of biological products is not caused by production errors: these molecules are intrinsically variable. Changes are inevitable during the process of producing biologics: changes in scale, innovations seeking efficiency, and new production sites. Changes for Remicade (37 changes reported to the regulator), Embrel (22 changes), Humira (20 changes) and Mabthera (6 changes) ranged from different providers of cell culture media, through new purification methods, to new manufacturing centers.[19]
The FDA has recognized that original producers still have been unable to precisely measure structural differences caused by production changes,[11] and such changes are accepted by regulatory entities without new clinical trials. Thus doctors obviously have been prescribing nonidentical versions of the same drug for years.
Regulatory agencies have established that product quality data should be compared before and after a change, that routine and continuous analyses should be conducted on batches, processes should be monitored and checked, and stability should be characterized and analyzed in comparison with historical data to ensure that batch variability and process changes have no adverse effects on safety or efficacy.[3] Very limited data from such studies are in the public domain. However, to request approval of a biosimilar, producers are required to submit analytical comparability and clinical information that is much more extensive and in-depth than the comparability reports submitted by original producing companies regarding changes in the production process after regulatory approval.[3]
A study by Sandoz Biopharmaceuticals provided examples of acceptable variations in various products on the market between 2007 and 2010, with different expiration dates. Batches of Aranesp (darbepoetin alfa) showed different sialylation rates and isoforms by expiration date. In commercial batches of Rituxan/Mabthera (rituximab) with expiration dates from September 2007 to October 2011, differences were detected even in antibody dependent cellular cytotoxicity activity. Enbrel showed different glycosylation profiles between batches with expiration dates at the end of 2009 and other batches with later expiration dates. Yet all three drugs stayed on the market without labelling amendments indicating that these changes did not produce clinical alterations and therefore were acceptable to health authorities.[20]
In contrast, there are also examples of apparently perfect analytical comparability of products that produced clinical differences when used on patients. This occurred in over 200 patients treated with erythropoietin who experienced red cell aplasia after changes in the formula of the original brand, Eprex. Human albumin was replaced by polysorbate 80, which reduced erythropoietin stability and reacted with uncoated rubber stoppers in prefilled syringes in which it was suppled. This caused leaching that added complex-forming substances that enhanced immunogenicity. After uncoated stoppers were replaced with ones less prone to leaching, red cell aplasia again became very rare.[21] This event shows that studies of product packaging may be even more important than large comparative clinical trials to assess immunogenicity of biosimilars.
How similar must a biosimilar be to the original product? Obinutuzumab, with negative results in B-cell lymphoma, but positive results with undifferentiated lymphomas, is a genetically modified anti-CD20 antibody to enhance FcγRIIIa receptor interaction. As a result, its activity against antibody-dependent cytotoxic cells, and particularly certain lines of B cells, is 100 times greater than that of the wild antibody. It has been demonstrated that this antibody is more effective than rituximab in eradicating malignant B cells in chronic lymphocytic leukemia.[22] The difference (~20%) between the first biosimilar infliximab and its original reference product may seem insignificant, especially when comparison is made by applying a target cell line that has been genetically modified to make it very sensitive to anti-tumor necrosis factor.[22]
6. Biosimilars work: There is mounting evidence of financial benefits without significant toxicity or loss of efficacy.
The World Oncology Forum highlighted the need for effective yet accessible cancer drugs in its call to world leaders to fight cancer.[23] The appeal has obliged many organizations, including WHO, to address this issue.[24]
Despite increasing debate, the first generation of biosimilars (related mainly to human protein therapy) has been extensively used in Europe since 2006.[25] The social value of a biosimilar depends on several variables: cost, the medical community’s assessment, who is making the political decisions, regulatory agency policies and pharmacoeconomic analyses.
Omnitrope, a somatropin biosimilar, was the first to receive EMA approval in 2006 and its use increased access to treatment.[26] Aapro published a cost–efficiency analysis of a filgrastim biosimilar for reducing chemotherapy-induced febrile neutropenia in the five countries where it is most used (Germany, France, Italy, Spain and the United Kingdom).[26] If 10,000 hypothetical cancer patients changed their treatment to the biosimilar for 4 days, it would save €3.9 million, and 347 and 132 additional patients could be treated with rituximab and trastuzumab, respectively. A 14-day treatment could generate savings of €13.7 million and permit additional treatment of 1213 and 461 patients with rituximab and trastuzumab, respectively.[26] Another study published by this group estimated savings of €146 million and an additional 12,913, 5117 and 4908 patients treated with rituximab, bevacizumab and trastuzumab, respectively, if 100,000 hypothetical patients had received erythropoietin biosimilar instead of the original.[27]
A recent meta-analysis of 58 clinical trials and more than 12,000 patients that also included pharmacovigilance data on biosimilars found no evidence of safety concerns related to switching between originals and biosimilars.[28]
Remsima, an infliximab biosimilar that entered the European market in 2013 (now approved in 72 countries and launched in 50 countries worldwide), has produced significant price reductions in Scandinavian countries, especially Norway. In February 2014 it was 39% cheaper than the original infliximab; in 2015, the difference was 72%.[29] In Denmark, Remsima occupy 96.2% of the market within a few months after its introduction, not only for new patients but also for those treated previously with the original. The EU Consortium of Individual Regulators’ position is that switching patients to and from biosimilars is safe.[30]
Savings associated with the use of biosimilars allow more patients access to biological therapies and contribute to changing medical practice, above all in low-income countries that could benefit from an increase in biotech treatments.
A NATIONAL BIOSIMILAR STRATEGY SHOULD BE GUIDED BY A SINGLE CRITERION: MAXIMIZE HEALTH IMPACT
From an ethical perspective, the main criterion for making public health decisions about a strategy for biosimilars should be population health impact, under conditions of equity, not exclusion.
The regulatory context usually has a dual effect on new products’ impacts on population health. On the one hand, a lack of regulations can be dangerous because it incentivizes low quality. On the other hand, a highly restrictive regulatory context could reduce health impacts by raising prices (without increasing quality) and thus limit population access.[31]
The problem we now face is that of a growing pharmacopeia with increasing numbers of biological products for multiple or long-term use in chronic diseases, with increasing costs, not so much because of intrinsic manufacturing costs, but because of monopoly effects and marketing. Under current conditions, it is impossible to meet the two goals of modern effective therapy and universal population coverage.
When will we reach a balance between safety and efficacy, on the one hand, and access on the other, in order to maximize public health impact? This problem is both legal and scientific. Countries should strengthen their manufacturing capacities and create their own strategies to tackle this complex issue in order to protect their populations and provide them with necessary medications.[31,32]
The issue is especially complex for biological products. Before biotechnology, once pharmaceutical patents expired, generic versions of the same quality were allowed on the market. In the USA, the Hatch–Waxman Law reduced regulatory barriers to generics, waiving requirements for repeating clinical trials if physical and chemical comparability could be proven.
It is not so simple, however, for biological products, since they are obtained through processes that introduce a variety of contaminants and differences among batches, even with the same producer. Biosimilars are comparable in theory to generics; although they are not exactly generics, (since there is no guarantee that the molecular structure is identical to the original as with generics from chemical synthesis) nor are they strictly new products, since they rely on a large volume of pre-established science regarding the type of molecule, its molecular targets and mechanisms of action.
The study of the effect of a monoclonal antibody on its receptor does not start from scratch with each new product. The theoretical guidelines of frequentist inferential statistics do not explicitly include prior knowledge in their analytical resources for assessing clinical comparability trials. Bayesian statistics, however, does include such prior knowledge, in the form of probability functions, in an analytical strategy based on totality of evidence, a concept that has been mentioned in many regulatory documents but not put into practice.[33]
The new panorama with biotech drugs requires a focus on universal therapeutic coverage, political will, intelligent management capabilities, and a strong scientific component. Clear goals need to be set and evaluated according to their impact on health indicators. Meaningful results from clinical trials and technical and economic viability of the manufacturing operation are nothing but intermediate steps toward the more important end goal, improving population health.
Analytical characterization, comparative pharmacokinetics and close postregistration pharmacovigilance components are complementary tools to improve estimation and assessment of the risk implicit in deciding to allow a given product entry into medical practice. Continued progress in analytical technologies suggests that a combination of these would provide better evidence than preregistration trials for demonstrating therapeutic equivalence. Such an evidence strategy would cost society less, and public health would be better served.