ABSTRACT
Sickle cell anemia is the most common hereditary disease in Cuba. On average, 1 in 33 Cubans is a carrier of this severe hemolytic anemia that can cause early death. In early 1980, its incidence in Cuba was calculated at 1 in 1600 births. In 1982, the Cuban public health system established the Sickle Cell Anemia Prevention Program, which aims to prevent the disease through identification of carrier couples and antenatal diagnosis of fetuses with disease-associated genotypes. In 1982–2018, hemoglobin genotypes were tested in 4,847,239 pregnant women. Of these, 168,865 (3.5%) were found to be carriers or to have sickle cell disease. During the same period, 8180 at-risk couples were identified, of whom 79.2% agreed to an antenatal study for detection of the sickle cell gene in the fetus. Among fetuses diagnosed, 20.1% had the SS genotype, the most clinically severe; 76.2% of the associated couples decided to interrupt the pregnancy. This program has resulted in a 3-fold reduction in prevalence of sickle cell disease in Cuba, a
10-fold reduction in the number of infants born with it each year, and a 16-year average increase in life expectancy of sickle cell disease patients of both sexes. Key contributors to these results have been universal screening of pregnant women in primary care, installation of diagnostic laboratories in every province, genetic counseling for couples, testing of fetal DNA (allowing couples to decide whether to continue the pregnancy if the fetus tests positive for the disease) and guaranteed multidisciplinary clinical care for patients. The Cuban experience shows that a middle-income country can mitigate the impact of a genetic disease through a universal preventive program based in primary care, which also pays particular attention to afflicted patients.
KEYWORDS Sickle cell anemia, sickle cell disease, sickle cell disorders, hemolytic anemia, sickle cell trait, sickle cell hemoglobin C disease, HbS disease, prevention, antenatal screening, preventive health services, Cuba
INTRODUCTION
Sickle cell disease (SCD) is a monogenic hereditary disease—a mutation in a single gene is sufficient to cause the disease, which originates in a point mutation in the alleles of the beta-hemoglobin gene. SCD affects most organs, and is characterized by abnormal erythrocytes that lose water and cations under low oxygen conditions due to hemoglobin S polymerization. Changes in the expression of cell adhesion molecules occur, small blood vessels become blocked, and severe hemolytic anemia occurs, characterized by vaso-occlusive crises with tissue ischemia and episodes of severe pain. Crises can be severe and lead to death at an early age.[1]
This recessive autosomal disease is caused by SS, CC and SC homozygotic genotypes. Patients with the SS genotype have the most severe form of SCD. Other allele combinations occur: the AA genotype in healthy individuals, and the AS and AC genotypes in carriers. When both individuals in a couple are asymptomatic carriers, each pregnancy has a 25% chance of producing a child with SCD.[1,2]
The number of children born with SCD in Latin America annually is estimated at about 6000.[3] Caribbean countries have a high SCD prevalence at birth, (0.26–0.65%)[2] second only to that in Sub-Saharan Africa, which had an estimated 230,000 cases at birth in 2010.[4] In Cuba the prevalence of SCD at birth is 0.02%, the lowest among Caribbean countries.[2] Although there are no substantiating statistics, clinical practice suggests that SCD is the most common monogenic hereditary disease in Cuba.
The genetic ancestry of Cuba’s population is a mix, representing—in order of relative frequency—Europeans, Africans and indigenous Americans. A 2014 study showed that on average, 20% of Cubans’ genetic information comes from African ancestors. In populations of African origin, the frequency of the S allele is 3%–18%. The magnitude of the African genetic contribution, regardless of skin color, explains the high frequency of the S allele in Cuba’s population.[2,5]
IMPORTANCE This article shows how a middle-income country can reduce sickle cell disease prevalence and increase patient life expectancy through a national, universal preventive program based in primary health care, which also provides multidisciplinary medical followup to children born with the disease.
By 1982, an average of 100 infants with SCD were being born annually in Cuba. This prompted the Cuban health system to establish an SCD prevention program, operating in a network that included primary care settings, hospitals and research institutions, as well as medical genetics laboratories. The Sickle Cell Anemia Prevention Program was designed to screen pregnant women and couples to detect carriers, and, if detected, to conduct antenatal diagnosis of SCD genotypes in the fetus. In 1982–2006, laboratory testing used hemoglobin electrophoresis on equipment designed and patented by Cuban researchers at the National Medical Genetics Center (CNGM).[6,7] In 2006, this manual technology was replaced with semiautomatic multiparameter electrophoresis, using Hydrasys equipment (Sebia, France), installed in the national medical genetics network’s 17 laboratories (1 in each province and 2 in Havana).[8]
In the 37 years since the design and implementation of the Program, CNGM in Havana has coordinated and conducted the national SCD surveillance system (unless otherwise noted, data cited in this article are from the Program’s databases). CNGM manages Cuba’s hemoglobin electrophoresis laboratories, including purchase and distribution of reagents and supplies for diagnosis. It continually monitors Program operations and statistics and performs fetal DNA testing for antenatal diagnosis. Program design follows the population-wide hereditary disease control program model recommended by WHO.[9]
CUBA’S SICKLE CELL ANEMIA PREVENTION PROGRAM
Implementation Begun in Havana in 1982, the Program was rolled out to other provinces through 1986. The first efforts consisted of training staff, purchasing reagents and supplies, manufacturing electrophoresis equipment and developing genetic counseling protocols. Since its inception, the program has used the algorithm described below.
SICKLE CELL ANEMIA PREVENTION PROGRAM ALGORITHM FOR DIAGNOSIS AND GENETIC COUNSELING
At the first antenatal appointment, the woman’s family doctor offers to perform the hemoglobin electrophoresis test and suggests she attend a genetic risk assessment interview in the genetic counseling department of her community polyclinic. The test is performed before the end of the first trimester. If she is a carrier or has the disease, the diagnostic test is offered to her partner to identify his disease status (healthy, carrier, or with SCD).
Blood samples are transported from the community polyclinic to the municipal medical genetics department, and from there to the hemoglobin electrophoresis laboratory in the provincial genetics department. Results are sent to the genetic counselor, who shares them with the couple (or with the pregnant woman alone, if necessary). Couples in which both members are carriers of the S or C alleles, or who have the disease (AS, AC, SS, SC and CC genotypes) are offered genetic counseling by a clinical geneticist and antenatal diagnosis of SCD (from week 15 to week 22 of the pregnancy) with a fetal DNA study.
For this study, a sample of amniotic fluid is sent to the molecular genetics laboratory at the National Medical Genetics Center, and the result is returned to the province of origin. The couple attends another genetic counseling session, in which information is provided, including the choice of therapeutic abortion for fetuses who will become sick as infants. For couples who decide to continue the pregnancy, hemoglobin electrophoresis is performed on the newborn to confirm diagnosis, and clinical monitoring in a specialized hematology department is begun, continuing followup over time.[10,11]
Genetic counseling provides education about SCD, its causes, consequences, management and prevention, and helps couples make independent choices about their child’s future during the pregnancy if the DNA study shows the fetus has an SCD-related genotypes.
In 1987, the Program reached 79% of pregnant women, and in 1989, more than 90%.[6,10,12] In the period 1995–2018, hemoglobin electrophoresis testing was performed on 98% of pregnant women in Cuba.
In 2007, digital databases were introduced to register hemoglobin electrophoresis results in medical genetics departments in community-based polyclinics, municipalities and provinces. This reduced the number of hemoglobin electrophoresis procedures performed, avoiding repeated tests on multiparous pregnant women whose results were already known. This allowed some pregnant women to avoid uncomfortable blood draws, optimized procedures and reduced costs by decreasing the number of samples drawn, stored, transported and processed. Since 2007, Cuba has saved US$4 (in reagents and supplies) for each repeat test avoided, thus reducing the annual cost to about US$500,000 (Program administrative data).
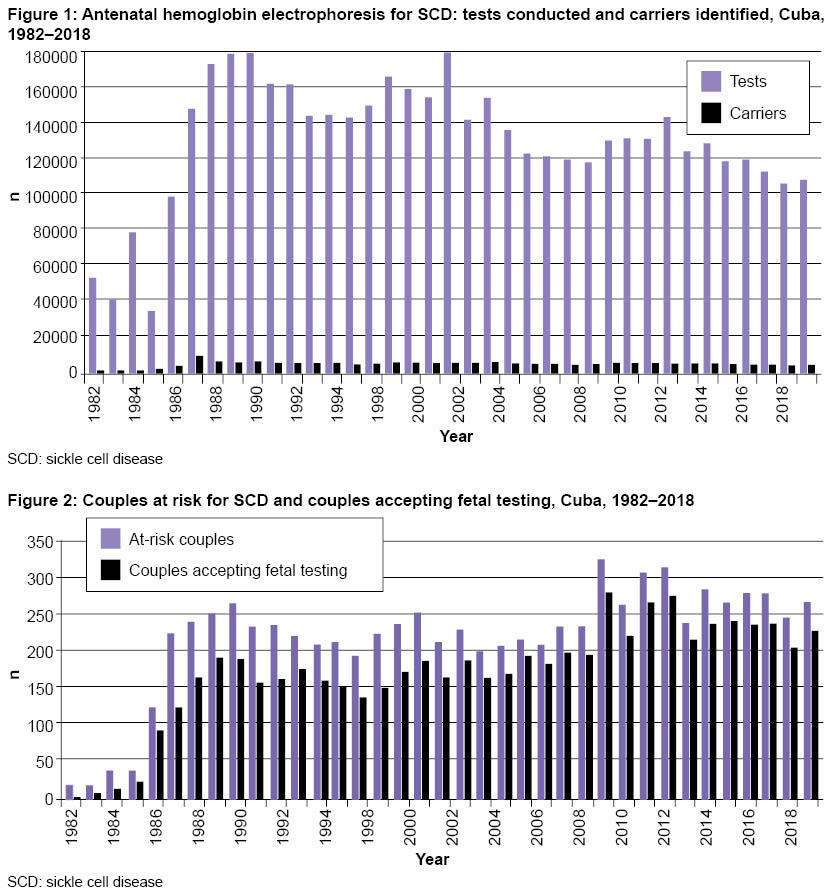
Carrier screening and genetic counseling From January 1982 through December 2018, hemoglobin genotypes were studied in 4,847,239 pregnant women (hemoglobin AA: normal; AS and AC: carriers; SS, SC and CC: SCD). Of these women, 3.5% were diagnosed as carriers or as having SCD because they had one or both variants of hemoglobin S or C (Figure 1). The most common genotype among carriers was AS, with an average frequency of 3.2% in the 37 years since Program inception. Next was the AC genotype, at 0.6%. In pregnant women with SCD, the SS and SC genotypes both occurred at a frequency of 0.02%.
Because SCD has an inherited recessive autosomal pattern, it is important to diagnose carrier status in both parents. The hemoglobin test was conducted on 143,626 men, 85.1% of those who needed to be tested. Holguín Province had the lowest percentage of men tested in Cuba (72.1%), and Havana had the highest (92.3%). The most common reasons why male partners were not tested were: refusal to accept the possibility of being carriers, failure to acknowledge paternity, living apart from the pregnant woman, and lack of interest in being diagnosed. According to standard Program protocols, information about reasons was collected in questionnaires. The data indicate that educational strategies used in genetic counseling to help couples make independent decisions after learning of their diagnosis (as well as SCD causes, consequences, management and prevention) were not fully effective in actively involving male partners in the risk identification program. In 1982–2018, the Program identified 8180 at-risk couples, and of these, 6475 (79.2%) agreed to antenatal diagnosis (Figure 2).
Antenatal diagnosis of SCD by fetal DNA testing Over the life of the Program, antenatal diagnosis using DNA testing identified 1299 fetuses with SCD-associated genotypes (SS, CC or SC), representing 20.1% of all tests. When a fetus tested positive, the couple was offered genetic counseling to help them make an independent decision about whether to continue the pregnancy. Of the couples (or women) facing the most severe forms (SS or SC genotypes), 76.2% chose termination. By contrast, those whose fetuses were CC genotype, which develop less severe forms of SCD, rarely chose to terminate. After birth, all SCD children are monitored and given special care by the Program.
In antenatal DNA studies performed on 3303 fetuses since 2004 through 2018, the most common carrier genotype was AS (1417 cases; 42.9%). Less common were genotypes AA (931 cases; 28.2%), SS (455 cases; 13.8%), AC (270 cases; 8.2%), SC (202 cases; 6.1%) and CC (12 cases; 0.4%).
The SS genotype occurred most frequently in the provinces of Cienfuegos (17%), Santiago de Cuba (15.3%), Holguín (14.9%), Havana (14.9%), Sancti Spíritus (14.9%) and Ciego de Ávila (14.8%). The SC genotype occurred most frequently in the provinces of Ciego de Ávila (11.1%), Villa Clara (9.7%), Sancti Spíritus (8.5%), Guantánamo (7.9%), Santiago de Cuba (7.7%) and Matanzas (7.1%).
 Since 2014, with the Program testing 99% of pregnant women, an average of 10 infants with SCD have been born each year, most of them due to couples’ informed decision to continue the pregnancy. Other reasons for this, which to some extent reveal weaknesses in the Program—but also respect the independence of individuals’ decision-making—were the mother’s refusal of SCD or carrier-status testing or not knowing the carrier or disease status of the male partner.
Since 2014, with the Program testing 99% of pregnant women, an average of 10 infants with SCD have been born each year, most of them due to couples’ informed decision to continue the pregnancy. Other reasons for this, which to some extent reveal weaknesses in the Program—but also respect the independence of individuals’ decision-making—were the mother’s refusal of SCD or carrier-status testing or not knowing the carrier or disease status of the male partner.
SCD carrier prevalence in pregnancy and disease incidence SCD carrier prevalence was calculated by number of pregnant women tested who had hemoglobin AS or AC. Fetal disease incidence was estimated by number of fetuses with SCD-associated genotypes over total pregnancies tested. Prevalence at birth was taken to be the number of infants born with SCD genotypes among all live births.
Among pregnant women studied in 37 years, 3.5% were diagnosed as SCD carriers, a rate that varied in different provinces, depending on genetic ancestry composition, particularly the percentage of African-origin genes in the population.[5] In regions with greater percentages of black or mestizo residents, the frequency of pregnant carriers and incidence of fetuses with SCD-associated genotypes was also higher. Santiago de Cuba and Guantánamo Provinces had the highest carrier prevalence, 6.3% and 5.3% of pregnant women tested, respectively, while in the central provinces of Villa Clara, Sancti Spíritus and Ciego de Ávila (where European-origin genetic makeup is more frequent), prevalence was 1.7%.
Knowing differences in carrier prevalence and disease incidence permitted the Program to develop priorities and strategies for each territory, including number of genetic counselors needed in each community genetics department, number of multidisciplinary teams for patient care, and cost estimates for maintaining the Program and related health care services.
During the Program’s existence, SCD prevalence at birth dropped from 1 in 1600 births in the 1980s to 1 in 5000 in 2018. The gradual decrease in disease incidence was confirmed through the Program registration system and clinical registry of genetic diseases. Rigorous monitoring was performed at provincial medical genetics centers, which used hemoglobin electrophoresis to detect carrier couples. Quality control of these tests and fetal DNA studies throughout Cuba were performed at CNGM.
SCD mortality in Cuba From 1987 to 2018, there were 1252 deaths from SCD. Life expectancy since Program implementation has shown a sustained increase. At the end of the 1980s, average age at death for a person with SCD was 28 years; during the 1990s, this rose to 32 years, then to 37 years in 2000–2009 and 44 years in 2010–2018. In Villa Clara Province, survival increased to 53 years in 2010–2018. Average age at death was slightly higher in females, as it was for national all-cause mortality rate. The 16-year increase in survival of patients with SCD (1987–2018) is the result of actions to address SCD through the prevention program, ranging from reducing the number of infants born with the most severe disease genotypes to continuous improvement in clinical attention to patients with SCD. Specialized followup clinical care is provided to children and all those with SCD in every Cuban province, in teams led by hematologists. This includes folic acid administration throughout life, oral prophylactic penicillin from 3 months through 5 years of age, and a daily dose of hydroxyurea (15 mg/kg).[11,13]
Despite the Program’s satisfactory results, deficiencies did occur in primary health care, where detailed, rigorous analysis of data obtained through Program protocols was not always performed. Such analysis would have allowed assessment and improvement of the Program’s organizational processes.
In addition, the Program did not achieve a successful communications strategy to increase public awareness of the Program’s SCD prevention and management services, and to encourage all pregnant women and their partners to agree to testing and antenatal fetal diagnosis.
CARIBBEAN NETWORK OF RESEARCHERS ON SICKLE CELL DISEASE AND THALASSEMIA (CAREST)
SCD is the genetic disease for which patterns are most similar among Caribbean countries. It was brought to the area through forced migration of more than 12 million Africans during the transatlantic slave trade.[14] The first report of SCD in the medical literature was made in the USA, when a young dental student from Grenada was diagnosed.[15] Carrier prevalence ranges from 13.8% and 13.6% in St. Lucia and Jamaica, respectively, to 10% in Martinique and French Guiana.[1] The country reporting the lowest percentage of carriers is Cuba, with 3.5%.[1]
Cuba was the first Caribbean country to implement nationwide antenatal screening to identify at-risk couples and provide therapeutic abortion upon the couple’s request which is not subject to legal exemptions or conditions.[12] Decisions about ending or continuing pregnancy are taken by both prospective parents, but women’s decisions are respected and protected.[16]
Jamaica was the first nation to demonstrate feasibility of neonatal screening for SCD and its impact on morbidity and mortality.[17] Neonatal screening programs exist in 7 other Caribbean countries, with coverage of over 96% in Trinidad and Tobago, 98% in Jamaica and Guadeloupe and 99% in Martinique; Grenada and St. Lucia report coverage of 79% and 45%, respectively.[1]
Before 2006, there were no reliable, consistent data on antenatal diagnosis of SCD in the region, with the exception of Jamaica, the French overseas territories, and Cuba. Starting in 2006, collaboration gradually began among research teams in various countries, now organized in the Caribbean Network of Researchers on Sickle Cell Disease and Thalassemia.[1]
As indicated in this paper, antenatal screening coverage in Cuba has been over 98.5% since 1999 and the number of infants born with SCD has decreased, while those living with SCD have increased survival rates. Cuba’s strategies have been shared with other CAREST researchers, whose countries nevertheless have adopted their own variants to address SCD, some of them quite different from the Cuban model. Some of these differences are attributable to religious, legal or ethical objections to abortion, or because they do not consider it necessary to offer the option of preventing SCD births, and restrict medical services to prophylactic antibiotic treatment and hydroxyurea to improve patient quality of life. This treatment is also used in Cuba in children with SCD,[11,13] when couples decide not to interrupt pregnancy when faced with an SCD diagnosis.
Notwithstanding differences in program strategies, systematic collaboration among multidisciplinary teams in the CAREST network encourages more detailed analysis of SCD’s origins and clinical and epidemiological characteristics, as well as the impact of programs and screening on morbidity, mortality and patient quality of life resulting from interventions carried out in the various countries.
CONCLUSIONS
Cuba’s Sickle Cell Anemia Prevention Program has been universally implemented throughout the country and integrated into public health strategies and actions. It has reduced SCD incidence and prevalence and, in followup of SCD children, has increased their survival rates through multidisciplinary clinical care.
Key factors in achieving these results have been universal screening in primary care to identify pregnant women who are carriers, diagnostic laboratories in every province, genetic counseling of at-risk couples and clinical management of SCD patients. The Cuban experience shows how a middle-income nation can mitigate a genetic disease’s impact through a national program that is universally implemented, free and accessible, based in primary health care.
The main weaknesses of the Program have been: poorly organized records for pregnant women who were carriers and those with SCD during its first 20 years, which led to unnecessary test repetition on multiparous pregnant women; an inadequate genetic counseling process that did not always motivate the male partner of a carrier to be tested; little analysis and assessment of the Program’s operations in polyclinics and municipalities for ongoing improvement of organizational procedures, and lack of a successful communications strategy for educating the general public.
References

- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet [Internet]. 2017Jul 15 [cited 2019 Feb 28];390(10091):311–23. Available from: https://doi.org/10.1016/S0140-6736(17)30193-9
- Knight-Madden J, Lee K, Elana G, Elenga N, Marcheco-Teruel B, Keshi N, et al. Newborn screening for sickle cell disease in the Caribbean: an update of the present situation and of the disease prevalence. Int J Neonat Screen [Internet]. 2019 [cited 2019 Feb 26];5(1):5–14. DOI:10.3390/ijns5010005. Available from: https://www.mdpi.com/2409-515X/5/1/5/
- Huttle A, Maestre G, Lantigua R, Green NS. Sickle cell in sickle cell disease in Latin America and the United States. Pediatr Blood Cancer [Internet]. 2015 [cited 2019 Aug 15];62:1131–6. Available from: https://www.researchgate.net/publication/273123730_Sickle_Cell_in_Sickle_Cell_Disease_in_Latin_America_and_the_United_States
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and popu lation estimates. Lancet [Internet]. 2013 Jan 12 [cited 2019 Feb 27];381(9861):142–51. Available from: http://www.sciencedirect.com/science/article/pii/S014067361261229X
- Marcheco-Teruel B, Parra EJ, Fuentes-Smith E, Salas A, Buttenschøn HN, Demontis D, et al. Cuba: exploring the history of admixture and the genetic basis of pigmentation using autosomal and uniparental markers. PLoS Genet [Internet]. 2014 Jul 24 [cited 2019 Feb 27];10(7):e1004488. DOI: 10.1371/journal.pgen.1004488. Available from: https://journals .plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004488
- Granda H, Gispert S, Dorticós A, Martín M, Cuadras Y, Calvo M, et al. Cuban programme for prevention of sickle cell disease. Lancet. 1991 Jan 19;337(8734):152–3.
- Heredero L, Granda H, Aguiar JA, Altland K. An economic high-speed electrophoretic screening system for hemoglobin S and other proteins. 1974 Feb 21;21(2):167–77.
- Díaz-Barroso LM, Cisneros-Zerquera H, Miguel-Morales M, Pérez-Diez de los Ríos G, Hernández-Martínez A, Uley-del Rosario G, et al. Técnicas electroforéticas en el HYDRASYS 2. Utilidad diagnóstica en diferentes enfermedades. Rev Cubana Hematol Inmunol Hemoter [Internet]. 2012 Jan–Mar [cited 2019 Feb 28];28(1). Available from: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892012000100011&lng=es. Spanish.
- World Health Organization. WHO Human Genetics Programme. (2000). Primary health care approaches for prevention and control of congenital and genetic disorders: report of a WHO meeting, Cairo, Egypt, 6–8 December 1999 [Internet]. Geneva: World Health Organization; 2000 [cited 2019 Feb 28]. 43 p. Available from: http://www.who.int/iris/handle/10665/66571
- Granda H, Gispert S, Martínez G, Gómez M, Ferreira R, Collazo T, et al. Results from a ref erence laboratory for prenatal diagnosis of sickle cell disorder in Cuba. Prenat Diagn. 1994 Aug;14(8):659–62.
- Svarch E, Marcheco-Teruel B, Machín-García S, Menéndez-Veitía A, Nordet-Carrera I, Arencibia-Núñez A, et al. La drepanocitosis en Cuba. Estudio en niños. Rev Cubana Hematol Inmunol Hemoter [Internet]. 2011 Jan–Mar [cited 2019 Feb 27];27(1):51–67. Available from: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892011000100005&lng=es. Spanish.
- Heredero-Baute L. Community-based program for the diagnosis and prevention of genetic disorders in Cuba. Twenty years of experience. Community Genet. 2004;7(2–3):130–6.
- Machín S, Svarch E, Agramonte O, Núñez A, Menéndez A, Hernández C, et al. Tratamiento con dosis moderadas de hidroxiurea en la drepanocitosis. Rev Cubana Hematol Inmunol Hemoter [Internet]. 2008 [cited 2019 Aug 15];24. Available from: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892008000200008&lng=es&nrm=iso&tlng=es. Spanish.
- Eltis D, Richardson D. 2015 Atlas of the Transatlantic Slave Trade. Connecticut: Yale University Press; 2015. 336 p.
- Herrick JB. Peculiar elongated and sickle-shaped red blood cell corpuscles in a case of severe anemia. 1910. Ann Intern Med (Chicago). 1910 Nov;6(5):517–21.
- Rojas I, Llamos A, Marcheco B. Aspectos éticos a tener en cuenta para el asesoramiento genético en los programas de diagnóstico prenatal de defectos congénitos en Cuba. Rev Cubana Genet Comunit [Internet]. 2012 [cited 2019 Aug 15]; 6(2):58–63. Available from: https://www.researchgate.net/publication/282355237. Spanish.
- Serjeant G, Serjeant BE, Forbes M, Hayes RJ, Higgs DR, Lehmann H. Haemoglobin gene frequencies in the Jamaican population: a study in 100,000 newborns. Br J Haematol. 1986 Oct;64(2):253–62.
THE AUTHOR
Beatriz Marcheco-Teruel (beatriz@infomed.sld.cu), physician specializing in medical genetics with a doctorate in medical sciences. Full professor, senior researcher and Director, National Medical Genetics Center, Havana, Cuba.
Submitted: March 29, 2019 Approved: July 17, 2019 Disclosures: None

