INTRODUCTION
Neurofibromatosis 1 (NF1) is one of the most common inheritable genetic disorders. It is characterized by formation of neurofibromas in skin, subcutaneous tissue, cranial nerves and nerves at the base of the spinal column. It is caused by various mutations in NF1 (a complex gene located on chromosome 17q11 2), resulting in reduced or absent action of the protein neurofibromin.[1,2]
NF1 diagnosis is based on seven established clinical criteria, each with strictly defined characteristics:[1,2] café au lait spots (CLS), freckles in places unexposed to sunlight, neurofibromas, Lisch nodules, optic glioma, characteristic bone lesions, and history of a first-degree relative with NF1. Any two of these are sufficient to establish NF1 diagnosis.[2,3–7]
Apart from the major clinical signs that constitute diagnostic criteria, there are others that contribute to the diagnosis, complications that occur in 33% of NF1 patients and are some of the most serious. These include plexiform neurofibromas and long bone pseudoarthrosis, as well as others that appear more frequently in NF1 than in the general population: learning disorders, scoliosis, mental retardation, cancer, central nervous system and spinal cord tumors, epilepsy and hypertension.[1–3,6] Individuals with NF1 may know their risk of transmitting the mutated gene, but have no way of knowing whether an affected child will develop complications.
In half the cases, NF1 is inherited (autosomal dominant pattern); in the other half, it arises from de novo mutation, the basis of which is still unknown. Thus its occurrence is unpredictable, precluding preventive measures. Having a child with de novo NF1 does not increase risk of having another affected child. In some studies, higher paternal age has been found in NF1 cases caused by new mutations, but this finding has not been confirmed.[1,2]
NF1 manifestations increase with age and not all are evident in early childhood; hence the argument that not all criteria are useful for diagnosing NF1 in children. Yet, over half the affected children have some clinical signs before their second birthday. CLS are a common early sign; usually present at birth, they can increase in size and number with age, so NF1 case finding should be based on identifying CLS.[3–8] By age 8 years, the diagnosis can be made with near certainty in almost all cases, and with virtually 100% certainty in persons aged ≥20 years. Pathogenic mutations of NF1 have full or nearly full penetrance in adults.[2]
NF1 is a disease with marked variability in expression.[3,4,6,8,9] Typically, treatment is only sought when complications appear in affected individuals or family members that create medical, aesthetic or functional problems. Apparently, concern is most acute among couples who have NF1 family history and are expecting a child.[2,8] Such variability and limited use of medical facilities may contribute to prevalence underestimates. Worldwide, reported NF1 prevalence is 1/3000 population,[6,8] with marked geographical differences: 1/7000 in northern Italy; 1/6000 in Canada; 1/4500 in Wales; 1/3000 in Finland, 1/2000 in New Zealand, and 1/1000 in Israel.[2] Active case finding can provide more accurate estimates of NF1 population prevalence.
NF1 prevalence in Cuba has not been estimated. The purpose of this study was to determine NF1 prevalence in a group of 9–11 year-old children in the province of Pinar del Río.
METHODS
Study type A descriptive cross-sectional study was carried out in all 14 municipalities in the province of Pinar del Río during May–June 2004.
Universe and study population The universe consisted of the 19,404 children aged 9–11 years (i.e., born between January 1, 1993 and December 31, 1994) living in the province at the time of the study. The study population consisted of 19,392 children assessed for NF1 in the survey.
Data collection To facilitate digitalization, a data collection form was designed, based on medical records used in Cuba’s medical genetics services. Content included general information, family history, family tree and findings on NF1-specific physical examination (Appendix 1, available online at www.medicc.org/mediccreview/orraca.pdf).
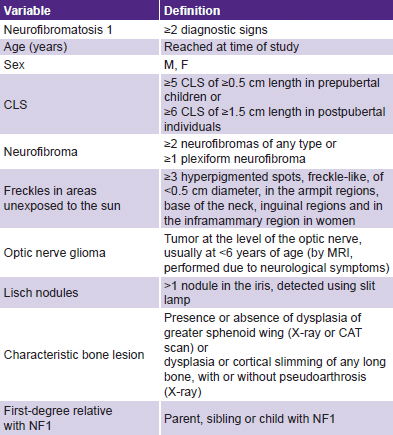
Variables (see Table 1)
Table 1: Variables
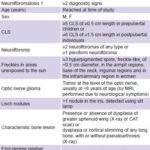
CLS: café au lait spots / NF1: neurofibromatosis 1
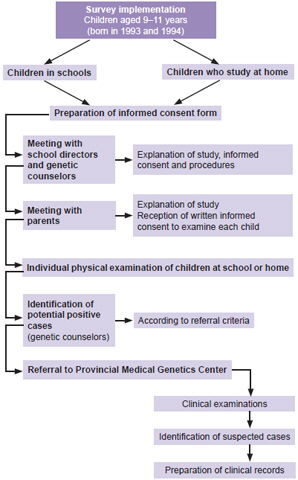
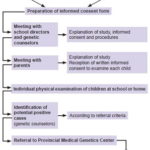
Procedures The study was conducted in two phases: the first, a survey of the entire population aged 9–11 years by genetic counselors in the province’s schools to assess for NF1 criteria; the second, evaluation by clinical geneticists of children who met criteria and were referred to the Provincial Medical Genetics Center (CPGM, the Spanish acronym) (Figure 1), where a complete medical history was taken to confirm NF1 diagnosis.
In the first phase, genetic counselors with prior training in recognition of NF1 clinical signs carried out physical examination of children in the study population in their learning environments: 633 elementary schools (whether general or special education) and homes (where disabled children received lessons by teachers in the school system). Twelve children were absent from school and could not be examined.
Figure 1: Study flow chart
Assessment was aimed at detection of easily recognizable clinical signs, particularly those most frequent in the first years of life in persons with NF1. The following constituted criteria for CPGM referral for the second phase of the study:[1–3,6]
- ≥5 CLS of any size (including freckles of any size in areas unexposed to sunlight)
- ≥5 hypochromic spots of any size
- ≥3 CLS or hypochromic spots of any size in a child attending a special school (since some complications of NF1 are associated with various types of disabilities that may require special schooling)
- any tumor visible on the skin
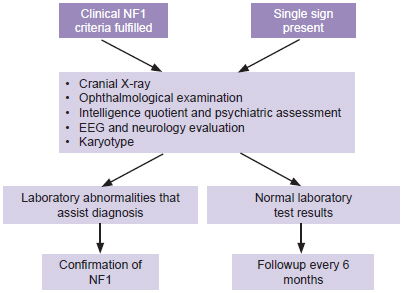
Following physical examination, children who met at least one of these criteria were referred to CPGM for assessment in the study’s second phase. As shown in Figure 2, this included a thorough history and physical examination, and preparation of a three-generation family tree. Peripheral blood was drawn for cytogenetic study to rule out chromosomal rearrangement as the cause of NF1.
Figure 2: Flow chart for children with CLS
Preparation of clinical record by history, physical examination and family tree.
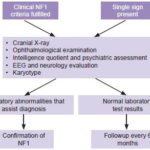
In cases with sufficient diagnostic signs to indicate NF1, first-degree relatives were also assessed to identify whether the mutation was inherited or de novo. Because of limited provincial access to MRI, optic nerve glioma was not among the signs explicitly sought, which did not preclude its identification if the patient had undergone MRI for other reasons.
Data management and analysis A database was created to store and manage study information with safeguards to protect patient identities. Access 2003 software run on Windows was used to develop the Management System for Patients with NF1 (SIGEPANEURO, the Spanish acronym).
EPI-INFO 2010 was used to create frequency distributions of variables and summarize results as percentages, presented in tables and figures. Prevalence was calculated for NF1 and its diagnostic signs (without confidence intervals because the study group constituted almost the entire universe).
Ethical considerations The protocol was formulated within a priority research line of Cuba’s Ministry of Public Health, and was approved by the CPGM ethics committee. All necessary safeguards were implemented to guarantee participant anonymity, and parents or guardians provided written informed consent.
Cuba’s public health policy and the organization of genetic services facilitated this research: our approach to the scientific problem was considered feasible and appropriate, and resources needed for the study were made available. The research did not generate conflicts of interest.
RESULTS
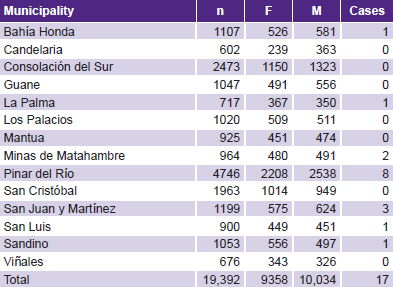
The surveyed population constituted 99.9% of the universe, 9358 girls and 10,034 boys (Table 2). After physical examination, 200 children were referred to CPGM for assessment; 17 of these children had clinical features consistent with NF1. temperature
CLS, freckles in areas unexposed to sunlight, characteristic bone lesions, history of NF1 in a first-degree relative, Lisch nodules, and peripheral neurofibromas were the diagnostic criteria seen, in order of frequency (Table 3). Of the surveyed children, 200 (1%) had CLS, 17 of whom (0.1%) also had hypochromic spots. In 11 children, CLS were disseminated over the whole body and in a single case they were countless, but seen only on the trunk. In 5 cases, CLS were predominantly located on the trunk, limbs and neck, but could be counted (5–19 in each location). Only two children had skin tumors.
In 12 of the 14 cases with freckles, they were located in both axillae; in 2 of them, both inguinal regions were also affected. One case had freckles only in the left inguinal region, and the other in the left axilla.
Table 2: Surveyed children by sex, municipality and NF1 cases

Table 3: Diagnostic signs in children with NF1 (n = 17)

X: present / CLS: café au lait spots / NF1: neurofibromatosis 1
Characteristic bone lesions were seen in 41.2% of NF1 patients. Two had long bone thinning and dysplasia (one of the right femur and the other of the right tibia); five had dysplasia of the greater sphenoid wings of the ocular orbit. Two children had peripheral neurofibroma; none had plexiform neurofibroma.
Other clinical abnormalities were detected in some children, among them signs known to accompany NF1, such as stunting (two cases), macrocephaly (four), learning disorders (five), mild mental retardation (one), epilepsy (three) and early puberty (one). Four children had undergone MRI for other reasons and in two of them, optic nerve gliomas were found, one unilateral and the other bilateral.
NF1 prevalence was 1 in 1340 children (7/9386) in the 1993 cohort and 1 in 1001 (10/10,006) in the 1994 cohort, for an overall prevalence of 1 in 1141 or 87.7 per 100,000 (17/19,392). Seven of those affected originated in an inherited mutation, the rest were the result of new mutations. The de novo/inherited ratio was 1.4. Only 4 of the 17 children diagnosed with NF1 were receiving specialized care (dermatology), none of them by clinical geneticists.
DISCUSSION
A large number of children were referred for clinical genetics assessment, since CLS are not infrequent in the general population, up to three is normal in children and up to five in adults.[2] Approximately one tenth of the population has one or two CLS.[3] The presence of a large number of CLS is suggestive of NF1, but must be accompanied by at least one other diagnostic criterion to confirm diagnosis.
Other studies have explored diagnostic criteria and their value in children and find the same two most frequent signs as in this study: CLS and freckles.[6,10] The third most frequent sign, Lisch nodules, can be seen at ages 6–10 years[3,4,8,9] but not all researchers have the slit lamps needed for their detection.
Peripheral neurofibroma may appear in early childhood, but is unusual. Generally this type of tumor is seen between adolescence and the fourth decade of life.[7,11]
Two crucial NF1 criteria in childhood are the characteristic bone lesions and history of the disease in a first-degree relative. The typical bone lesion is cortical slimming of a long bone, often the tibia. Identification of dysplasia of the ocular orbit’s greater sphenoid wings requires radiological study, but is of great diagnostic value, as we found. Some bone lesions require imaging confirmation, which can be obtained through inexpensive and noninvasive studies (x-rays).[1–4,8,9,12]
Optic gliomas tend to appear around age three years in NF1 patients and may be accompanied by exophthalmos, acute vision loss or blindness.[2,13–15] Optic glioma incidence in NF1 is estimated at 15%; 30%–58% of these are astrocytomas, which may regress spontaneously. The children we studied were older than the usual age of onset for optic glioma, and that, plus the need for an MRI to detect them, led to our decision not to include optic glioma as a criterion for proceeding to the study’s phase two.
Authors of articles reporting prevalence have inferred that the range of NF1 incidence is 23–40 per 100,000 newborns (estimates based on prevalence in pediatric populations, in whom it is assumed that clinical signs found were present from birth).[10–12,16,17] The prevalence we found is higher than in most studies reviewed, which used different methodologies, such as post-mortem studies or assessment of patients spontaneously attending genetic services.[2,3,8,9,18–21]
This makes comparison difficult. Only Lammert used a methodology similar to ours, examining children in schools from six states in Germany in 2000–2001, and finding NF1 prevalence of 1 in 2996, lower than in our study. However, this was in a population of children aged six years, in whom perhaps the signs indicating medical genetics referral had not yet appeared. Furthermore, due to unavailability of diagnostic tests, only four cardinal criteria for referral were used,[22] compared to six in ours, offering a slightly greater possibility of diagnostic certainty.
We consider our prevalence estimate valid; that is, the risk of NF1 in the province is higher than that reported in some other parts of the world. Thanks to active casefinding, the study population covered almost the entire universe. This was possible because in Cuba it is very difficult for children of this age not to be enrolled in the education system. By law, all Cuban children attend primary school (1st through 6th grade, i.e. aged 5–12 years), which allowed extension of the study through age 11 years. If disability or a learning disorder prevents school attendance, children receive instruction at home, but they are still in the educational system, allowing researchers ready access. In addition, the study was carried out in ages when diagnostic signs should already be present, as they usually appear by age eight years.[2]
For these reasons, it is unlikely that prevalence was underestimated. Because of the specificity of confirmatory tests, it was probably not overestimated either. Therefore, this research was the first population-based clinical/epidemiological study in Cuba to examine a large number of persons to identify patients with NF1.
It is estimated that new mutations are found in 50% and have even been reported above that figure,[1–4,8–12] as observed in our research. However, thorough examination of the parents of affected children is a must, in order to rule out inherited mutations.
NF1 diagnosis and information on all affected persons in a geographic area are crucial inputs for counseling by medical genetics professionals. Presumably, increased NF1 awareness by patients and professionals will eventually reduce frequency of the disease and its avoidable complications. Prevention is only feasible if affected patients and their relatives know their risks and understand the range of preventive options, from pregnancy avoidance to interventions to reduce incidence or intensity of NF1 complications.
CONCLUSIONS
Active NF1 case finding in a population group confirmed a higher than expected prevalence, and that most cases are not detected through routine well-child examinations in primary care.