ABSTRACT
Spinocerebellar ataxia type 2 is a degenerative disease that causes physical disability and, ultimately, prostration and death. Globally, reported prevalence is around 3 cases per 100,000 population and Cuba has the world’s highest rates of the disease, affecting both patients and their at-risk descendants. In Holguín Province, which has the country’s highest concentration of cases, incidence is 4.4 per 100,000 population and prevalence is 40.2 per 100,000 population. In 2000, a specialized research center was established in that province. Supplied with the necessary equipment and human resources, the center conducted national multidisciplinary studies involving molecular biology, clinical care, epidemiology, psychology, clinical neurophysiology, imaging, clinical genetics and community medicine, among others. A training and continuing education program also raised scientific capacity. Priority was given to developing international collaborations for academic exchange and training of Cuban researchers.
Multiple results from research involving clinical and epidemiologic characterization of the disease, identification of biomarkers and therapeutic targets, genetic association studies, clinical trials and characterization of the disease’s preclinical stages have been introduced in care of patients and their at-risk descendants. This has been accomplished through various programs including personalized rehabilitation, predictive diagnosis and social services. These results have also been published in high-impact scientific journals and received national and international awards. Such an experience in the context of Cuba’s national health system—which is universal, free, accessible, comprehensive, prevention-oriented and with a record of international cooperation—demonstrates the possibility of providing quality care to affected families. Incorporating research findings into medical practice, with the resulting impact on patients’ health and wellbeing, is a practical example of translational medicine in Cuba.
KEYWORDS Spinocerebellar ataxia type 2, health services research, biomedical research. health care delivery, translational medicine, translational research, health equity, Cuba
INTRODUCTION
Spinocerebellar ataxias are hereditary degenerative diseases causing physical disability eventually leading to prostration and death. More than 45 molecular types have been described, most frequently spinocerebellar ataxias types 1, 2, 3 and 6. Fuller understanding of their etiology and pathogenesis requires highly specialized genomic, proteomic and metabolomic studies, as well as research with experimental models.[1,2]
Spinocerebellar ataxia type 2 (SCA2) is a hereditary, neurodegenerative, disabling disease with an autosomal dominant inheritance pattern. It follows a progressive course that affects motor functions related to coordination, gait, postural stability and speech. It produces cognitive and sleep disturbances, as well as dysautonomia. SCA2 inevitably leads to prostration and death, produced by degeneration of the cerebellum and other central and peripheral nervous system structures.[3–5]
SCA2 has low prevalence in most regions of the world, reported globally at 3 per 100,000 population. Cuba has the world’s highest concentration of SCA2 patients and at-risk descendants.[6,7] The disease is concentrated in the over one million inhabitants of Holguín Province, northeastern Cuba, where incidence rate is 4.4 and prevalence is 40.2 per 100,000 population. Confronting the disease has been a major challenge for Cuba, requiring enormous efforts by the public health system and government to acquire the necessary technology for molecular diagnosis programs; conduct research with scientific, clinical and social impact; and sustain a presence in the international scientific community. An extra challenge was to develop such advanced science in Holguin, nearly 800 km from Havana.
IMPORTANCE This article presents an example of a model for comprehensive management of ataxias that could be useful for other regions of the world. It demonstrates the application of ataxia research in services provided to Cuban patients, their families and descendants, highlighting the value of human resource development as well as international collaboration.
Cuba’s single public health system provides universal, free, accessible, comprehensive, prevention-oriented care, and has a tradition of collaboration with other countries. The system also prioritizes scientific research, technological development and innovation in different fields. It comprises several medical universities and 37 research centers known as Science and Technological Innovation Entities (ECIT).[8]
To meet the SCA2 challenge, the Ministry of Public Health initiated a project for the study of hereditary ataxias and in March 2000, the Center for the Research and Rehabilitation of Hereditary Ataxias (CIRAH) was established in Holguín City, the provincial capital.
From the beginning, CIRAH’s primary mission has been to develop scientific research and technological innovations aimed at finding therapies to modify the clinical course and severity of hereditary ataxias. CIRAH also provides medical care to improve the quality of life of patients and their families, and contributes to academic training and continuing education of researchers, technicians and other professionals in the field of hereditary ataxias and other movement disorders.
A little over four years after its founding, CIRAH attained ECIT status in recognition of its research results, accelerated training of human resources, development of scientific projects with important social impact, and application of specialized health care programs. The latter include comprehensive, personalized rehabilitation; molecular prenatal and presymptomatic diagnosis; and social services for affected families.[9]
The study and treatment of SCA2 have involved basic, clinical, epidemiologic and social research. This article describes and analyzes the program’s history, research impacts, medical practices, ethical implications, international cooperation, and family education; as well as public health policies, strategies and practices promoting social welfare and personalized care that have improved quality of life for affected families.
CUBA’S SCA2 PROJECT
In the study of SCA2, Cuba has reached top levels in terms of scientific development and productivity. The text describes how this has come about.
Research history The story begins with the first observations of increased numbers of patients and with various epidemiologic studies conducted since the late 1990s, continuing to the present. Among other aspects, these studies have estimated national SCA2 prevalence and incidence, described the disease’s evolution by stage in order of prevalence, and evaluated policy and strategy outcomes.
In the 1960s, Dr Rafael Estrada Pérez, then director of the Cuban Neurology and Neurosurgery Institute in Havana, noticed that most patients with hereditary ataxias came to that institution from Oriente Province, which at the time included the current provinces of Holguín, Las Tunas, Granma, Santiago de Cuba and Guantánamo.[10] In 1978, Vallés reported a prevalence of ataxias in that region of 48.4 per 100,000 population.[10] In 1977, Beguería studied 30 patients with autosomal dominant ataxia and found significant ocular movement alterations.[11]
In the 1980s, a hereditary ataxia study was conducted in the V.I. Lenin University Hospital in Holguín’s provincial capital, and collaboration with the University of Düsseldorf, Germany, was initiated. In 1981, Cordovés studied a group of ataxia patients in Tacajó, a health service area in Báguanos, Holguín Province, and as a result, estimated a population of more than 1000 affected individuals for the whole province.[12] In the late 1980s, Nodarse carried out nerve conduction and evoked-potentials studies in families with hereditary ataxias.[13] In 1989, Orozco described the clinical characteristics of patients with autosomal dominant cerebellar ataxia in Holguín and detected a significantly low concentration of dopamine in patients’ cerebrospinal fluid, associated with neuronal depletion in the substantia nigra.[14]
In 1990, Auburger began to study the “founder effect,” which occurs when specific mutations become more prevalent in the population of a determined geographic area, and argued that this phenomenon existed in Holguín, given the high prevalence of SCA2 in that province.[15] In November 1996, scientists mapped the gene associated with SCA2 and the mutation that causes it, which consists of an expanded cytosine-adenine-guanine (CAG) repeat sequence in the 5’ end of the ATXN2 gene.[16−18] This discovery led to an important group of molecular, clinical, genetic and neurophysiologic studies that facilitated identification of biomarkers and therapeutic targets, as well as development of clinical trials.
The late 1990s and early months of the year 2000 marked the beginning of a new stage in hereditary ataxia research, with the development of health care programs offering presymptomatic and prenatal diagnosis plus intensive personalized rehabilitation. In 2000, biomarkers were identified and CIRAH was established. A strategy was also initiated for training specialized human resources to develop new research in the fields of molecular biology, clinical epidemiology, neurophysiology and neurochemistry, among others. Major national and international collaboration projects were undertaken, generating increased numbers of publications in high-impact scientific journals.
Cuba is one of the countries contributing most to the body of knowledge about ataxias. Patient studies are ongoing, and characterization of the preclinical phase in at-risk mutation carriers has been completed. Several new studies of early rehabilitation are now under way. All this is particularly important, since identification and characterization of the mutation’s preclinical carriers offer early diagnosis and hence the possibility of early disease management through personalized clinical and psychological monitoring. This includes potential to apply various therapeutic options before disease onset, with a view to delaying appearance of the first motor symptoms.
Main health care outcomes and impact Health care services include multidisciplinary consultations at CIRAH and also in the community; personalized rehabilitation; and presymptomatic and prenatal diagnosis programs. The aim is to improve the quality of life of patients and their descendants at risk of becoming ill with the disease by applying medical and social measures that slow progression and onset. In this context, the goal is also to support decision-making and, in the case of predictive diagnosis programs, family planning in particular.
CIRAH’s programs are national in scope. Health care and research activities are carried out with patients and their descendants nationwide. Genetic diagnosis includes the molecular forms of spinocerebellar ataxias types 1, 2, 3, 6, 7, 8, and 17, as well as dentatorubral-pallidoluysian atrophy and Friedreich’s ataxia.
Programs with highest impact include presymptomatic[19] and prenatal[20] predictive diagnosis. In fact, CIRAH’s experience with this type of diagnosis is unique in the field of ataxias. These programs involving different specialists have been applied for more than 15 years. The protocol includes genetic counseling sessions, psychological and psychometric evaluations to determine if an individual is prepared to receive the diagnosis, and subsequent monitoring.[21,22] Research has shown that individuals’ levels of anxiety and depression decline considerably after receiving diagnosis, and no severe events (such as suicide) have been reported following ataxia diagnosis (something that occurs frequently with other types of degenerative disorders).[23,24] These outcomes benefited patients, their healthy relatives and asymptomatic carriers of the mutation. Moreover, these programs are distinguished for their ability to manage ethical dilemmas associated with this diagnosis.[25]
Overall, the prenatal diagnosis program has been requested by 201 couples, but has been necessary in only 108 cases of carriers who risked transmitting the mutation to their offspring. The presymptomatic diagnosis program has benefitted 1376 individuals, with 92% adherence and increased demand in the last 10 years (CIRAH administrative data).
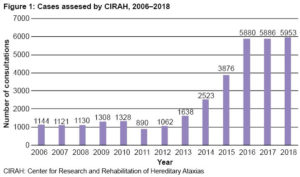
Clinical care includes multidisciplinary consultations with neurologists, psychologists, psychiatrists, internists, physiatrists, etc. The number of cases assessed annually has increased, particularly in recent years (Figure 1).
Main research outcomes and impact Various studies have been carried out over CIRAH’s nearly 20-year existence, beginning with the first epidemiologic investigations and molecular, neurophysiologic, clinical and neurochemical research. The primary objective of early investigations was identification of biomarkers and target variables for designing clinical trials and generating an SCA2 symptomatic treatment protocol. That strategy began earlier, in 1998, with an epidemiologic study in Holguín Province.[7] Since then, two more national studies have been conducted by CIRAH: one in 2003[6] and the other ongoing since 2017.
These national epidemiologic studies of hereditary ataxias are unprecedented globally: they have identified >160 families affected with some type of autosomal dominant ataxia, involving >700 patients and some 10,000 at-risk relatives. SCA2 is the molecular form with highest incidence and prevalence globally. In Cuba, it affects >100 families and altogether >500 patients, representing a proportion greater than 70% of all dominant ataxias.[6,7]
The findings informed social service provision for affected families, including walkers, canes and wheelchairs; housing and adaptive housing in the neediest cases; and rehabilitation in the community and at municipal and provincial hospitals. More than 800 patients have received physical rehabilitation, 70% of whom have shown improvement in cerebellar functions such as gait, balance, postural stability and speech. Frontal-executive functions, memory, attention and concentration improved in 75% of cases. Followup studies of patients receiving physical rehabilitation for five years showed slowed disease progression.[26−28] The advantages of physical rehabilitation during the disease’s preclinical stage has been recently demonstrated, opening the way for early therapeutic protocols to be put into place.[29]
Identification and localization of affected families—patients as well as asymptomatic carriers—enabled characterization of biomarkers of genetic damage (maximal saccade velocity, REM sleep without atonia, antisaccadic eye movements);[30−32] preclinical biomarkers (amplitude of sensitive evoked potentials, maximal saccade velocity, P40 component latency, central motor conduction time, percentage of REM sleep),[33−36] and progression biomarkers (amplitude of sensitive evoked potentials, periodic leg movements, maximal saccade velocity, percentage of REM sleep, etc.).[31,37,38] This also facilitated identification of therapeutic targets susceptible to eventual treatments or clinical trials.[4]
Identification and characterization of the abovementioned, primarily neurophysiologic, biomarkers enabled evaluation of the nervous system using various techniques. These studies over almost 30 years were conducted in >400 patients, 100 preclinical-stage cases and some 400 controls, and are part of CIRAH’s 40 research projects, a number of them with international collaboration. The electrophysiologic studies used most broadly in biomarker descriptions include electronystagmographic investigations. These have identified saccade slowing as a pathognomonic sign of the disease, endophenotypic marker, biomarker of progression and genetic damage, and preclinical biomarker.[30,34,38,39]
Principal study results are the definition and characterization of the prodromal stage of the disease, which precedes onset of the cerebellar syndrome.[33−37,40−42] These findings opened up a new phase, not only from a research point of view but in terms of early intervention as well. Also identified were clinical alterations––such as periodic leg movements during sleep, painful muscle contractions and cognitive disruption––that could be included as response variables in clinical trials.[5,31,32,41−43]
Molecular research was conducted in collaboration with scientists from Germany and the USA. This is the case of the SCA2 genetic modifiers study that identified the alpha 1 calcium channel subunit gene as a modifier of age onset variance and SCA2 severity.[44]
Additional molecular research topics have included a study of other genetic modifiers,[45−47] epigenetic factors, genetic predisposition, founder effect, molecular epidemiology, de novo mutations, the pathogenetic role of premutation alleles,[48−51] molecular characterization of Huntington’s chorea[52] and SCA3,[53] as well as studies of survival, genetic risk in at-risk descendants, and estimation of age at onset.[54−56]
Development of a transgenic animal model of SCA2 was a major scientific outcome that led to an important collaboration with the Havana-based Genetic Engineering and Biotechnology Center (CIGB).[57] Additional neurochemical research has been conducted on oxidative stress issues.[58,59]
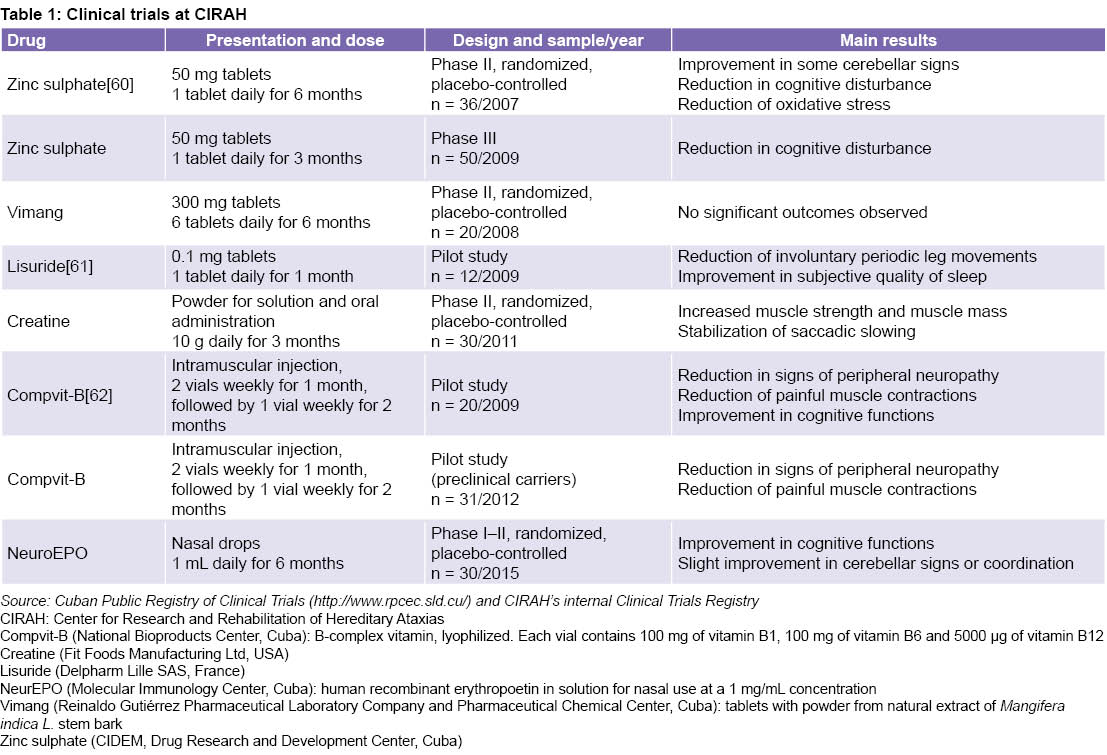
Other clinical trials involving nearly 200 patients and 31 preclinical individuals are shown in Table 1. Several of these results were published in international journals, facilitated doctoral training, and received national and international awards.
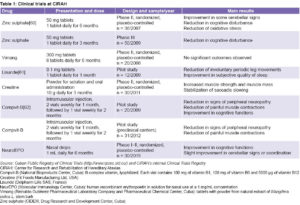
The first SCA2 treatment protocol was created, contributing to improvement in the quality of life for patients and preclinical carriers of the mutation.[4,29]
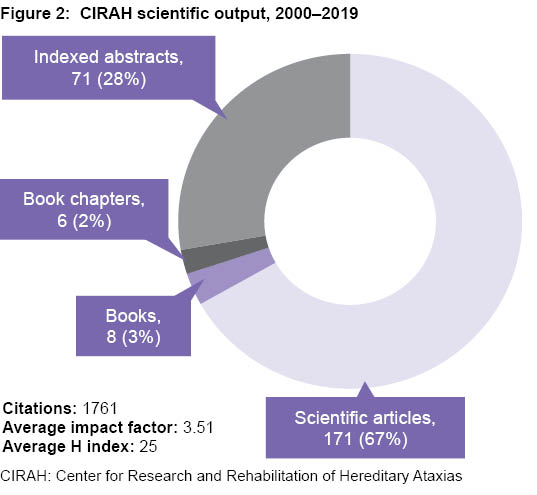
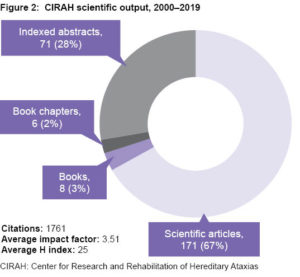
Scientific publications and awards At this writing, CIRAH’s work is represented in 256 publications, consisting of 8 books, 6 book chapters, 171 scientific articles and 71 peer-reviewed abstracts. Of these, 67% appear in Web of Science, 80% in SCOPUS and 96% in SciELO. These resulted in 1761 citations, an average publication impact factor of 3.51, and an average H index of 25 (Figure 2).
CIRAH’s scientific achievements have been recognized with more than 130 national and international awards. Nationally, CIRAH has won 29 Annual Health Awards from the Ministry of Public Health, 22 of these in the last 5 years, as well as 14 national awards from the Cuban Academy of Sciences. These numbers make CIRAH the Cuban institution with the highest number of awards per researcher and per PhD (1.4/researcher and 2.8/PhD), and the highest number of national awards to an institution located outside the nation’s capital.
CIRAH’s 57 international prizes include the Alexander von Humboldt Foundation’s Georg Forster Research Award (Germany), as well as several awards from the International Parkinson and Movement Disorder Society and the International Brain Research Organization.
Scientific potential and collaboration Some 83% of CIRAH researchers have master’s or doctoral degrees, 13 of them PhDs in various fields, including clinical neurophysiology, computer sciences, psychology, biology, microbiology, internal medicine, physical education and dentistry.
Nationally, CIRAH has also collaborated on epidemiologic studies with all of Cuba’s medical universities, as well as with the National Laboratory Animal Production Center, CIGB, Cuban Neuroscience Center, Comandante Manuel Fajardo Hospital, Pedro Kourí Tropical Medicine Institute, Neurology and Neurosurgery Institute, Molecular Immunology Center, National Clinical Trials Coordinating Center, Drug Research and Development Center, National Medical Genetics Center, and the Universities of Havana, Santa Clara and Holguín.
CIRAH has collaborated internationally on projects with institutions such as the German universities of Tubingen, Aachen and Frankfurt on electrophysiological and molecular analysis and imaging studies; the National Rehabilitation Institute of Mexico and National Autonomous University of Mexico on neurorehabilitation projects and detection of other molecular types of ataxia, such as SCA7; the National Medical Genetics Center of Venezuela; Neuroscience Center of Antioquía, Medellín, Colombia; and University of Victoria, Canada. In 2017, a research collaboration was established with the University of Chicago, USA.
Challenges and prospects One of CIRAH’s principal challenges is sustainability of research, health care and human resource training to deepen knowledge of the mechanisms triggering SCA2 and thus improve prevention and treatment strategies.
Our description of CIRAH as a model for application of scientific research to ataxia characterization and patient care also presents the new challenge of extending strategies to other Cuban institutions, fundamentally those associated with neurosciences and biotechnology. This would allow nationwide harmonization of treatment protocols, leading to a national initiative for early diagnosis and treatment of ataxias. CIRAH’s research and development model may be applicable as well in other institutions with similar scientific and health care profiles.
The experience of the Cuban ataxia project offers further evidence of the importance of translational medicine, defined as clinical application of research outcomes in the shortest possible time. This concept contrasts with the basic research paradigm in which search for and identification of new mutations may proceed without corresponding advances in effective treatments. Promoting such a paradigm shift represents a major challenge for Cuban and other researchers worldwide.
CONCLUSIONS
The fundamental reason for the Cuban SCA2 project led by CIRAH is the application of scientific research towards the goal of better understanding disease for earlier detection and treatment, and to eventually devise more effective treatment protocols. The project offers an example of introducing research results alongside development and application of comprehensive, multidisciplinary intervention programs with positive impacts and improved quality of life for individuals with the disease and those in its preclinical stage.
Over the past two decades, the project has included training and updating of skilled human resources, development of scientific leadership, and financial, material and scientific support obtained through both national and international collaboration.
In particular, the project’s and CIRAH’s own scientific development was led by researchers’ efforts, while international collaboration not only assisted with costly technology, but also provided an effective channel for sharing information and new knowledge. This approach facilitated development of strategies and concrete actions that ultimately made CIRAH a reference institution in the field of ataxias.
Conducting this cutting-edge research in a center far from the Cuban capital is also an indication of the national public health system’s priorities and scientists’ commitment to bring both scientific research and health care services to the places where health problems are found.
References

- Ashizawa T, Öz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol. 2018;14(10):590−605.
- Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res. 2018;7. pii: F1000 Faculty Rev-1781.
- Auburger GW. Spinocerebellar ataxia type 2. Handb Clin Neurol. 2012;103:423−36.
- Velázquez-Pérez LC, Rodríguez-Labrada R, Fernández-Ruiz J. Spinocerebellar ataxia type 2: clinicogenetic aspects, mechanistic insights, and management approaches. Front Neurol. 2017 Sep 11;8:472.
- Velázquez-Pérez L, Rodríguez-Labrada R, García-Rodríguez JC, Almaguer- Mederos LE, Cruz-Mariño T, Laffita-Mesa JM. A comprehensive review of spinocerebellar ataxia type 2 in Cuba. Cerebellum. 2011 Jun;10(2):184−98.
- Velázquez-Pérez L, Sánchez-Cruz G, Santos Falcón N, Almaguer Mederos LE, Escalona Batallan K, Rodríguez Labrada R, et al. Molecular epidemiology of spinocerebellar ataxias in Cuba: insights into SCA2 founder effect in Holguín. Neurosci Lett. 2009 Apr 24;454(2):157−60.
- Velázquez-Pérez L, Santos-Falcón N, García-Zaldívar R, Paneque-Herrera M, Hechavarría-Pupo R. Epidemiología de la ataxia hereditaria cubana. Rev Neurol. 2001;32(7):606−11. Spanish.
- Rojo Pérez N, Valenti Pérez C, Martínez Trujillo N, Morales Suárez I, Martínez Torres E, Fleitas Estévez I, et al. Ciencia e innovación tecnológica en la salud en Cuba: resultados en problemas seleccionados. Rev Panam Salud Pública [Internet]. 2018 Apr 24 [cited 2019 Apr 17];42:98‒108. Available from: https://www.scielosp.org/pdf/rpsp/2018.v42/e32/es. Spanish.
- Velázquez Pérez L, Rodríguez Labrada R, Sánchez Cruz G, Laffita Mesa JM, Almaguer Mederos L, Aguilera Rodríguez R, et al. Caracterización integral de la ataxia espinocerebelosa 2 en Cuba y su aplicación en proyectos de intervención. Rev Cub Salud Pública. 2011;37(3):230−44. Spanish.
- Vallés L, Estrada GL, Bastecherrea SL. Algunas formas de heredoataxia en una región de Cuba. Rev Neurol. 1978;27:163−76. Spanish.
- Beguería R. Alteraciones de la Motilidad Ocular en las Enfermedades Heredodegenerativas Espinocerebelosas Tipo Pierre Marie en la región del Norte de Oriente. Estudio prospec tivo de 30 enfermos [thesis]. [Havana]: Institute of Neurology and Neurosurgery (CU); 1977. 182 p. Spanish.
- Cordovés SR. Estudio clínico epidemiológico de las enfermedades heredodegenerativas espinocerebelosas tipo Pierre Marie en un área de salud de la provincia de Holguín [thesis]. [Holguín]: “Vladimir Ilich Lenin” Provincial Teaching Hospital; 1981. Spanish.
- Nodarse A, Orozco DG, Gutiérrez S, Coutin P. Clinical electrophysiological correlation in patients with hereditary ataxia and asymptomatic relatives. EEG Clin Neurophysiol. 1987;66:573.
- Orozco G, Estrada R, Perry TL, Araña J, Fernández R, González-Quevedo A, et al. Dominantly inherited olivopontocerebellar atrophy from eastern Cuba. Clinical, neuropathological and biochemical findings. J Neurol Sci. 1989 Oct;93(1):37−50.
- Auburger G, Díaz GO, Capote RF, Sánchez SG, Pérez MP, del Cueto ME, et al. Autosomal dominant ataxia: genetic evidence for locus heterogeneity from a Cuban founder-effect population. Am J Hum Genet. 1990 Jun;46(6):1163–77.
- Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat Scientific potential and collaboration Some 83% of CIRAH researchers have master’s or doctoral degrees, 13 of them PhDs in various fields, including clinical neurophysiology, computer sciences, psychology, biology, microbiology, internal medicine, physical education and dentistry.
- Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996 Nov;14(3):277−84.
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expaned CAG/ glutamine repeats. Nature Genet. 1996 Nov;14(3):285−91.
- Cruz-Mariño T, Velázquez- Pérez L, González-Zaldívar Y, Aguilera-Rodríguez R, Velázquez-Santos M, Vázquez-Mojena Y, et al. Cuban adolescents requesting presymptomatic testing for spinocerebellar ataxia type 2. ISRN Genetics [Internet]. 2013 [cited 2019 Apr 5];2013. Available from: https://www.hindawi.com/journals/isrn/2013/837202/
- Cruz-Mariño T, Velázquez-Pérez L, González-Zaldívar Y, Aguilera-Rodríguez R, Velázquez-Santos M, Vázquez-Mojena Y, et al. Couples at risk for spinocerebellar ataxia type 2: the Cuban prenatal diagnosis experience. J Community Genet. 2013 Oct;4(4):451–60.
- Cruz-Mariño T, Vázquez-Mojena Y, Velázquez-Pérez L, González-Zaldívar Y, Aguilera-Rodríguez R, Vázquez-Santos M, et al. SCA2 predictive testing in Cuba: challenging concepts and protocol evolution. J Community Genet. 2015;6(3):265−73.
- Cruz Mariño T, Velázquez Pérez L, González Zaldívar Y, Aguilera-Rodríguez R, Velázquez-Santos M, Vázquez-Mojena Y, et al. The Cuban program for predictive testing of SCA2: 11 years and 768 individuals to learn from. Clin Genet. 2013 Jun;83(6):518−24.
- Paneque HM, Prieto AL, Reynaldo RR, Cruz MT, Santos FN, Almaguer ML, et al. Psychological aspects of presymptomatic diagnosis of spinocerebellar ataxia type 2 in Cuba. Community Genet. 2007;10(3):132−9.
- Paneque M, Lemos C, Sousa A, Velázquez L, Fleming M, Sequeiros J. Role of the disease in the psychological impact of pre-symptomatic testing for SCA2 and FAP ATTRV30M: experience with the disease, kinship and gender of the transmitting parent. J Genet Couns. 2009 Oct;18(5);483−93.
- Cruz Mariño T, Armiñán RR, Cedeño HJ, Mesa JM, Zaldívar YG, Rodríguez RA, et al. Ethical dilemmas in genetic testing: examples from the Cuban program for predictive diagnosis of hereditary ataxias. J Genet Couns. 2011 Jun;20(3):241−8.
- Pérez-Ávila I, Fernández JA, Martínez-Góngora E, Ochoa Mastrapa R, Velázquez-Manresa MG. [Effects of a physical training program on quantitative neurological indices in mild stage type 2 spinocerebellar ataxia patients]. Rev Neurol. 2004 Nov 16–30;39(10):907−10. Spanish.
- Rodríguez JC, Velázquez L, Sánchez G, Almaguer L, Almaguer D, García JC, et al. Evaluación de la restauración neurológica en pacientes con ataxia SCA2 cubana. Plast & Rest Neurol. 2008 Jan–Dec;7(1–2):13−8. Spanish.
- Rodríguez-Díaz JC, Velázquez-Pérez L, Rodríguez-Labrada R, Aguilera R, Laffita D, Canales N, et al. Neurorehabilitation therapy in spinocerebellar ataxia type 2: a 24-week, rater-blinded, randomized, controlled trial. Mov Disord. 2018 Sep;33(9):1481−7.
- Velázquez-Pérez L, Rodríguez-Díaz JC, Rodríguez-Labrada R, Medrano-Montero J, Aguilera Cruz AB, Reynaldo-Cejas L, et al. Neurorehabilitation improves the motor features in prodromal SCA2: a randomized, controlled trial. Mov Disord. 2019 Apr 8. DOI: 10.1002/mds.27676.
- Velázquez-Pérez L, Seifried C, Santos-Falcón N, Abele M, Ziemann U, Almaguer LE, et al. Saccade velocity is controlled by polyglutamine size in spinocerebellar ataxia 2. Ann Neurol. 2004 Sep;56(3):444−7.
- Velázquez-Pérez L, Voss U, Rodríguez-Labrada R, Auburger G, Canales Ochoa N, Sánchez Cruz G, et al. Sleep disorders in spinocerebellar ataxia type 2 patients. Neurodegener Dis. 2011;8(6):447−54.
- Rodríguez-Labrada R, Velázquez-Pérez L, Aguilera-Rodríguez R, Seifried-Oberschmidt C, Seifried-Oberschmidt C, Peña-Acosta A, et al. Executive deficit in spinocerebellar ataxia type 2 is related to expanded CAG repeats: evidence from antisaccadic eye movements. Brain Cogn. 2014 Nov;91:28−34.
- Velázquez-Pérez L, Rodríguez-Labrada R, Canales-Ochoa N, Medrano-Montero J, Sánchez-Cruz G, Aguilera-Rodríguez R, et al. Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol. 2014 May;13(5):482−9.
- Velázquez-Pérez L, Seifried C, Abele M, Wirjatijasa F, Rodríguez-Labrada R, Santos-Falcón N, et al. Saccade velocity is reduced in presymptomatic spinocerebellar ataxia type 2. Clin Neurophysiol. 2009 Mar;120(3):632−5.
- Rodríguez-Labrada R, Velázquez-Pérez L, Canales Ochoa N, Galicia Polo L, Haro Valencia R, Sánchez Cruz G, et al. Subtle rapid eye movement sleep abnormalities in presymptomatic spinocerebellar ataxia type 2 gene carriers. Mov Disord. 2011 Feb 1;26(2):347−50.
- Velázquez-Pérez L, Rodríguez-Labrada R, Torres-Vega R, Ortega-Sánchez R, Medrano-Montero J, González-Piña R, et al. Progression of corticospinal tract dysfunction in pre-ataxic spinocerebellar ataxia type 2: a two-years follow-up TMS study. Clin Neurophysiol. 2018 May;129(5):895−900.
- Velázquez Pérez L, Sánchez Cruz G, Canales Ochoa N, Rodríguez Labrada R, Rodríguez Díaz J, Almaguer Mederos L, et al. Electrophysiological features in patients and presymptomatic relatives with spinocerebellar ataxia type 2. J Neurol Sci. 2007 Dec 15;263(1−2):158−64.
- Rodríguez-Labrada R, Velázquez-Pérez L, Auburger G, Ziemann U, Canales-Ochoa N, Medrano-Montero J, et al. Spinocerebellar ataxia type 2: measures of saccade changes improve power for clinical trials. Mov Disord. 2016 Apr;31(4):570−8.
- Rodríguez-Labrada R, Vázquez-Mojena Y, Canales-Ochoa N, Medrano-Montero J, Velázquez-Pérez L. Heritability of saccadic eye movements in spinocerebellar ataxia type 2: insights into an endophenotype marker. Cerebellum Ataxias. 2017 Dec 19;4:19.
- Velázquez-Pérez L, Rodríguez-Labrada R, Laffita-Mesa JM. Prodromal spinocerebellar ataxia type 2: prospects for early interventions and ethical challenges. Mov Disord. 2017 May;32(5):708−18.
- Velázquez-Pérez L, Rodríguez-Labrada R, Cruz-Rivas EM, Fernández-Ruiz J, Vaca-Palomares I, Lilia-Campins J, et al. Comprehensive study of early features in spinocerebellar ataxia 2: delineating the prodromal stage of the disease. 2014 Oct;13(5):568−79.
- Rodríguez-Labrada R, Velázquez-Pérez L, Ortega-Sánchez R, Peña-Acosta A, Vázquez-Mojena Y, Canales-Ochoa N, et al. Insights into cognitive decline in spinocerebellar Ataxia type 2: a P300 event-related brain potential study. Cerebellum Ataxias. 2019 Mar 4;6:3.
- Rodríguez-Labrada R, Velázquez-Pérez L, Seigfried C, Canales-Ochoa N, Auburger G, Medrano-Montero J, et al. Saccadic latency is prolonged in spinocerebellar ataxia type 2 and correlates with the frontal-executive dysfunctions. J Neurol Sci. 2011 Jul 15;306(1–2):103−7.
- Pulst SM, Santos N, Wang D, Yang H, Huynh D, Velázquez L, et al. Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A channel modifies age onset. 2005 Oct;128(Pt 10):2297−303.
- Almaguer-Mederos LE, Almaguer-Gotay D, Aguilera-Rodríguez R, González-Zaldívar Y, Cuello-Almarales D, Laffita-Mesa J, et al. Association of glutathione S-transferase omega polymorphism and spinocerebellar ataxia type 2. J Neurol Sci. 2017 Jan 15;372:324−8.
- Simon DK, Zheng K, Velázquez L, Santos N, Almaguer L, Figueroa P, et al. Mitochondrial complex I gene variant associated with early age at onset in spinocerebellar ataxia type 2. Arch Neurol. 2007 Jul;64(7):1042−4.
- Almaguer-Mederos LE, Aguilera-Rodríguez R, Cuello-Almarales D, Almaguer-Gotay D, González-Zaldívar Y, Vázquez-Mojena Y, et al. Normal ATXN2 alleles influences on the age at onset in spinocerebellar ataxia type 2. Mov Disord. 2017 Sep;32(9):1329−30.
- Laffita-Mesa JM, Bauer PO, Kourí V, Peña Serrano L, Roskams J, Almaguer Gotay D, et al. Epigenetics DNA methylation in the core ataxin-2 gene promoter: novel physiological and pathological implications. Hum Genet. 2012 Apr;131(4):625−38.
- Laffita-Mesa JM, Rodríguez Pupo JM, Moreno Sera R, Vázquez-Mojena Y, Kourí V, Laguna-Salvia L, et al. De novo mutations in ataxin-2 gene and ALS risk. PLoS One. 2013 Aug 6;8(8):e70560.
- Laffita-Mesa JM, Velázquez-Pérez LC, Santos Falcón N, Cruz-Mariño T, González-Saldívar Y, Vázquez-Mojena Y, et al. Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles. Eur J Hum Genet. 2012 Jan;20(1):41−9.
- Almaguer-Mederos LE, Mesa JML, González-Zaldívar Y, Almaguer-Gotay D, Cuello-Almarales D, Aguilera-Rodríguez R, et al. Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in spinocerebellar ataxia type 2. Clin Genet. 2018 Oct;94(3−4):346−50.
- Vázquez-Mojena Y, Laguna-Salvia L, Laffita-Mesa JM, González-Zaldívar Y, Almaguer-Mederos LE, Rodríguez-Labrada R, et al. Genetic features of Huntington disease in Cuban population: implications for phenotype, epidemiology and predictive testing. J Neurol Sci. 2013 Dec 15;335(1–2):101−4.
- González-Zaldívar Y, Vázquez-Mojena Y, Laffita-Mesa JM, Almaguer-Mederos LE, Rodríguez-Labrada R, Sánchez-Cruz G, et al. Epidemiological, clinical, and molecular characterization of Cuban families with spinocerebellar ataxia type 3/Machado-Joseph disease. Cerebellum Ataxias. 2015 Feb 21;2:1.
- Almaguer-Mederos LE, Proenza CL, Almira YR, Batallán KE, Falcón NS, Góngora EM, et al. Age-dependent risks in genetics counseling for spinocerebellar ataxia type 2. Clin Genet. 2008 Dec;74(6):571–3.
- Almaguer-Mederos LE, Falcón NS, Almira YR, Zaldívar YG, Almarales DC, Góngora EM, et al. Estimation of the age at onset in spinocerebellar ataxia type 2 Cuban patients by survival analysis. Clin Genet. 2010 Aug;78(2):169−74.
- Almaguer-Mederos LE, Aguilera Rodríguez R, González Zaldívar Y, Almaguer Gotay D, Cuello Almarales D, Laffita Mesa J, et al. Estimation of survival in spinocerebellar ataxia type 2 Cuban patients. Clin Genet. 2013 Mar;83(3):293−4.
- Aguiar J, Fernández J, Aguilar A, Mendoza Y, Vázquez M, Suárez J, et al. Characterization of a new transgenic mouse model of the spi nocerebellar ataxia type 2. Biotecnol Aplicada. 2006;23(1):54−9.
- Almaguer-Gotay D, Almaguer-Mederos LE, Aguilera-Rodríguez R, Estupiñán-Rodríguez A, González-Zaldívar Y, Cuello-Almarales D, et al. Role of glutathione S-transferases in the spinocerebellar ataxia type 2 clinical phenotype. J Neurol Sci. 2014 Jun 15;341(1−2):41−5.
- Almaguer-Gotay D, Almaguer-Mederos L, Aguilera-Rodríguez R, Rodríguez-Labrada R, Cuello-Almarales D, Estupiñán-Domínguez A, et al. Spinocerebellar ataxia type 2 is associated to the extracellular loss of superoxide dismutase but not catalase activity. Front Neurol. 2017 Jun 13;8:276.
- Velázquez-Pérez L, Rodríguez-Chanfrau J, García-Rodríguez JC, Sánchez-Cruz G, Aguilera-Rodríguez R, Rodríguez-Labrada R, et al. Oral zinc sulphate supplementation for six months in SCA2 patients: a randomized, double-blind, placebo-controlled trial. Neurochem Res. 2011 Oct;36(10):1793–800.
- Velázquez-Pérez L, Rodríguez-Labrada R, Álvarez-González L, Aguilera-Rodríguez R, Álvarez Sánchez M, Canales-Ochoa N, et al. Lisuride reduces involuntary periodic leg movements in spinocerebellar ataxia type 2 patients. Cerebellum. 2012 Dec;11(4):1051−6.
- Rodríguez-Labrada R, González Gay OT, Velázquez-Pérez L, Aguilera Rodríguez R, Canales Ochoa N, Coira Moreno Y, et al. Preliminary evaluation of the effect of Compvit-B on memory and learning processes in patients with SCA2. Rev Cubana Invest Bioméd. 2014;33(2):140‒9.
THE AUTHORS
Roberto Rodríguez-Labrada, microbiologist and neurophysiologist with a doctorate in health sciences. Youth member, Cuban Academy of Sciences. Full professor, associate researcher and head of research, Center for Research and Rehabilitation of Hereditary Ataxias (CIRAH), Holguín, Cuba.
Jacqueline Medrano-Montero, orthodontist with a doctorate in medical sciences. Professor, senior researcher and director, CIRAH, Holguín, Cuba.
Luis Velázquez-Pérez (Corresponding author: velazq63@gmail.com), neurologist and clinical neurophysiologist with a doctorate in medical sciences and an advanced doctorate in science. Distinguished Member and president of the Cuban Academy of Sciences, Havana. Full professor and visiting professor at the University of Holguín and founding director of the Pan American Network of Hereditary Ataxias. Senior researcher, CIRAH, Holguín, Cuba
Submitted: March 29, 2019 Approved: July 17, 2019 Disclosures: None