INTRODUCTION
Pre-eclampsia (PE) is a pregnancy-induced hypertensive disorder, accompanied by edema, proteinuria or both, occurring at >20 weeks gestation. It is primarily affects nulliparous women with no history of cardiovascular or kidney problems and is reversible post partum.[1]
PE is a leading cause of morbidity and mortality in pregnancy worldwide, accounting for 3–8% of pregnancy complications, including perinatal death, preterm birth and intrauterine growth retardation.[1–5] Together with eclampsia, PE is responsible for 10–15% of the over 500,000 deaths from pregnancy-related disorders globally (99% of these in developing countries).[1,3] In Cuba, PE is among the leading causes of maternal death and there has been an apparent rise in the proportion of maternal mortality related to hypertensive disorders of pregnancy since 2009, but numbers are small: 4 maternal deaths in 2012, a rate of 3.2 per 100,000 live births.[6,7]
The precise etiology of PE is unknown. However, it is considered a complex, multfactoral disease, in which individual manifestations depend on the interaction of two or more maternal genes with the fetal genotype and with environmental factors.[8] The main risk factors associated with PE and eclampsia are maternal age (risk is higher at the extremes of age, <18 or >35 years), nulliparity, malnutrition, poverty, low educational level, multiple pregnancy, molar pregnancy, diabetes and lupus erythematosus.[8] Other factors associated with elevated PE risk are s protein deficiency, anticardiolipin antibodies, pre-pregnancy obesity, and genetic factors such as family history (mothers, sisters and daughters of PE patients have greater frequency of the disorder).[6,8]
Some studies have identified chromosome regions and candidate genes whose variants are related to greater PE susceptibility, but their results have not been replicated consistently across populations.[9,10] The following have been shown to increase PE susceptibility in different settings:[9]
- polymorphism of the methylenetetrahydrofolate reductase enzyme gene, a flavoprotein involved in homocysteine remethylation;
- a common mutation leading to substitution of a glutamine by an arginine at position 506 in the gene encoding for factor V Leiden;
- angiotensinogen gene polymorphisms;
- common polymorphisms in the gene encoding for lipoprotein lipase; and
- the NOS3 gene, located at region 7q36, which encodes for endothelial nitric oxide synthase.
The US National Center for Biotechnology Information genome database includes other genes that may predispose to PE: the TGF-a gene; the alpha-adducin gene, which encodes a protein associated with calmodulin, whose mutations generate changes in blood pressure regulation in rats; the tachykinin receptor 1 gene, related to neurokinin B, a substance implicated in vascular regulation in PE; the gene for annexin 4, a placental anticoagulant protein; adhesion molecule genes; and many others that could be related in some way to PE physiopathology.[8,9]
Evidence has accumulated on the respective genetic and environmental contributions to PE predisposition,[8,11,12] but the potential interaction of the two should also be considered. This is fundamental if the goal is proper risk assessment for personalized preventive genetic counseling and more effective prenatal care to prevent pregnancy complications. Although there have been Cuban PE risk-factor studies,[13–15] no studies have examined the effect of genetic and environmental components and their interaction on PE risk. This may explain why there are no risk reference tables for PE attributable to genetic predisposition (signaled by family history), environmental risk factors, or their interaction. This problem leads us to ask: Does interaction between a predisposing genome and environmental risk factors contribute to PE risk beyond the effect of the individual factors acting independently?
Hence the hypothesis is put forward that the interaction of a woman’s genetic predisposition to PE and adverse environmental factors could increase risk of PE more than if genetic and environmental factors were acting separately. Ours is the first Cuban study to address this hypothesis.
METHODS
Type of study and subjects This exploratory phase of an analytical hospital-based case-control study used data for deliveries from January 2007 through December 2009 at the Eusebio Hernández University Hospital, a maternity hospital in Havana. The 312-bed hospital is located in the municipality of Marianao and serves a population of approximately 89,000 women of childbearing age, primarily from Playa, La Lisa and Marianao municipalities. It is the provincial referral center for care of neonates weighing <1500 g. The hospital has more than 4000 births annually.
Study universe This consisted of the 124 pregnant women admitted to the hospital’s hypertension and pregnancy service, their records culled from the hospital database. Of these, the 80 patients who met the inclusion criteria below were selected for the study population.
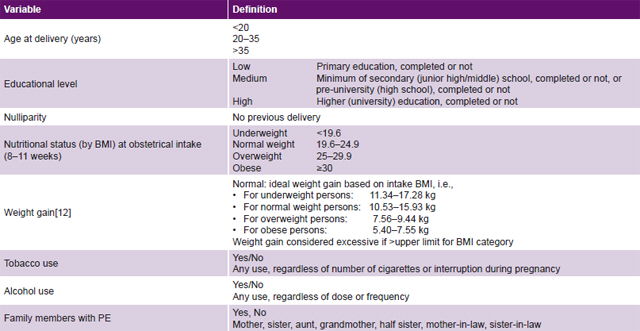
Table 1: Study variables
BMI: body mass index PE: pre-eclampsia
Clinical and laboratory PE diagnostic criteria Systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg, associated with proteinuria (excretion of ≥0.3 g protein in 24-hour urine), both detected in a previously healthy pregnant woman at >20 weeks’ gestation.[12]
Inclusion criteria Residence in La Lisa municipality (for convenience; two of the authors work in La Lisa); delivery at Eusebio Hernández University Hospital within the study period; clinical and laboratory diagnosis of PE made by specialists of the hypertension and pregnancy service.
Exclusion criteria Other clinical forms of hypertension in pregnancy, such as gestational hypertension, chronic hypertension, chronic hypertension with superimposed PE, and eclampsia.
Control group A sample of 160 controls (2 per case) was selected using density sampling and matched by year of admission, age and polyclinic geographic catchment area to which they belonged at the time of study.
Study variables See Table 1.
Data collection Data from physical examination at time of admission were used and in-depth interviews were conducted with both cases and controls for pedigree construction, using an instrument designed for the study and validated by an expert group of two obstetrician/gynecologists specializing in hypertensive disorders of pregnancy, two clinical geneticists and a biostatistician.
The instrument was used to obtain solicited data, following an interview guide. If the patient had limited knowledge of her family history, other relatives were interviewed. Data were recorded in Microsoft Excel 2007, and analyzed using SPSS v. 20.0 and INFOSTAT statistical software.
Statistical analysis
Environmental risk factors The Pearson chi-square test for independence and homogeneity and Fisher exact test were used, with statistical significance level set at α = 0.05. For factors for which distribution was statistically significant between cases and controls, phi (φ) correlation coefficient and odds ratio (OR) were calculated as measures of association strength. The null hypothesis for a difference of proportions of two independent samples was tested, using MICROSTAT statistical software. All non-genome-dependent factors were considered environmental for this analysis.
Genetic factors Familial aggregation was studied for each degree of kinship, using a case-control approach and always excluding the proband (pregnant subject for whom pedigree was constructed).[16]
The heritability coefficient (h²) was determined from the correlation between first-degree (sisters) and second-degree (half sisters) relatives, estimated by the proportion of concordance of the proband with each group.
The following formula was used to calculate h²:
h2 = 4 x (correlation between full siblings minus correlation between half siblings)
Since heritability is a proportion, the maximum value is 1; if the value is >0, both genetic and environmental factors are involved in disease occurrence. If h2 is >0.75, genetic involvement is considerable.[17]
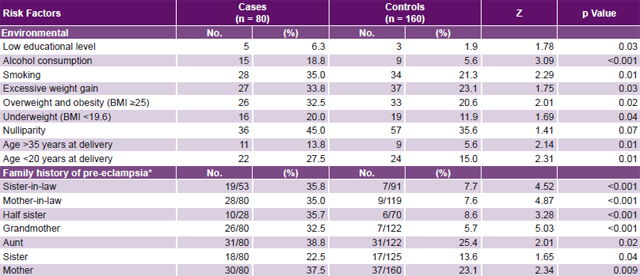
Table 2: Distribution of environmental risk factors and family history of pre-eclampsia in cases and controls
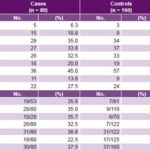
* For cases and controls, the quotient represents percentage of relatives who had pre-eclampsia among all relatives reported for that kinship degree (denominator signifies number of relatives of kinship degree who were asked about presence of pre-eclampsia; numerator is number verified).
BMI: body mass index
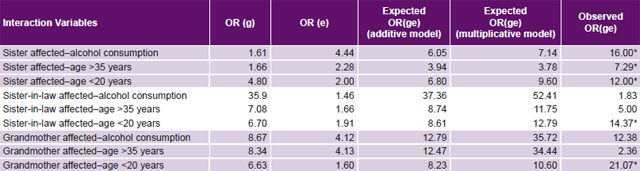
Study of genome–environment interaction A case-control study was designed to calculate the OR for genome–environment interaction. Analysis of PE family history by kinship degree was considered a proxy for the genome.[17] The three environmental risk factors and three genetic risk factors (kinship degree) with the highest ORs for PE were selected for analysis. Interaction was considered important if the observed OR for the effect of genetic and environmental factors together ORge, (OR gene–environment interaction) was greater than expected in both additive (ORg + ORe) and multiplicative (ORg x ORe) models.[17]
Ethics Written informed consent was obtained from patients and relatives involved in the study and all information collected was kept confidential. The study was approved by the research ethics committee of the National Medical Genetics Center.
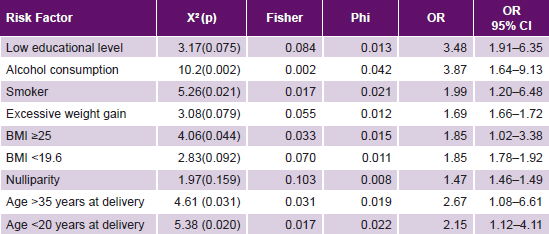
Study of environmental risk factors The most frequent non-genome dependent factors in both cases and controls were nulliparity, smoking, excessive weight gain, and BMI ≥25. All these environmental risk factors studied were significantly more frequent in cases than in controls, with the exception of nulliparity (Table 2). Of these, case control analysis found five to be statistically significant risk factors for PE, with greatest ORs being alcohol consumption, age >35 years and age <20 years at delivery (Table 3).
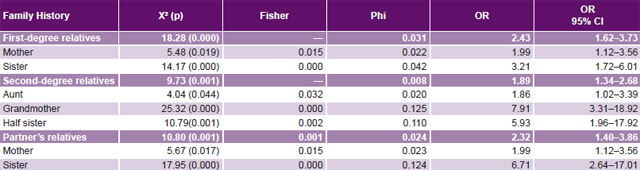
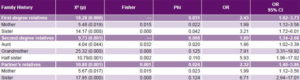
Study of genetic factors Family history of PE was more common in cases than in controls, most frequently in aunts, mothers and sisters-in-law, and least frequently in sisters (Table 2). Familial clustering of PE was observed in relatives of cases compared to those of controls (p < 0.05). Among the environmental risk factors studied, alcohol showed the strongest effect on pre-eclampsia risk (OR 3.87, 95% CI 1.64–9.13). Familial pre-eclampsia clustering was observed; risk was increased for both first-degree (OR 2.43, 95% CI 1.62–3.73) and second-degree (OR 1.89, 95% CI 1.34–2.68) relatives as well as for husband’s relatives (OR 2.32, 95% CI 1.40–3.86). Based on OR, family histories conferring greatest PE risks were those with PE grandmothers (7.91), sisters-in-law (6.71) and half sisters (5.93) (Table 4).
As the proportion of shared genes increased, so did PE probability in the family history, significantly more so in cases than in controls.
Overall, higher correlation values were observed for family history than for environmental factors (Tables 3 and 4). The heritability coefficient (h2) was 0.24, indicating involvement of both genetic and environmental factors in PE.
Table 3: Case-control analysis of hypothesized environmental risk factors for PE
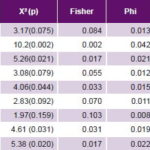
BMI: body mass index PE: pre-eclampsia
Table 4: Case-control analysis of familial PE clustering by degree of kinship
PE: pre-eclampsia
Table 5: Interactions between most significant environmental and family history PE risk factors
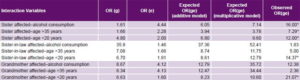
ORg: odds ratio for predisposing genome by family history alone ORe: odds ratio for environmental risk factors alone ORge: odds ratio for gene–environment interaction PE: pre-eclampsia * Statistically significant interaction
Study of genome–environment interaction Of the nine interactions, five were statistically significant for both the additive and multiplicative models (Table 5). Having an affected sister and one of the three environmental risk factors with the highest significant ORs (alcohol consumption, aged >35 years and aged < 20 years at delivery) amplified interaction impact on risk. Furthermore, when aged < 20 years interacted with genetic susceptibility, the result was always statistically significant. When a woman with a sister with PE history also used alcohol, PE risk increased by 16 times (ORge = 16.00), compared to the expected OR for an independent effect for either factor (OR for history of sister with PE = 1.61 and OR for alcohol consumption = 4.44; additive model expected OR 6.05, multiplicative 7.44).
When a grandmother with PE was identified and the pregnant woman was aged < 20 years at delivery, the OR for the interaction was 21.07 (greater than the OR of each of these factors separately), and if the pregnant woman was aged < 20 years and her husband’s sister had PE, then ORge = 14.37 (Table 5).
DISCUSSION
Analysis of environmental risk factors In this study, alcohol consumption was the environmental factor associated with greatest PE risk, almost quadruple that of non-drinkers. In Mexico, Morgan-Ortiz found an even stronger association between alcohol use and PE risk (OR 5.77, 95% CI: 1.48–22.53).[18] However, the finding is not seen in all populations. In Thailand, Fang found no association between alcohol consumption and PE.[19] Differences among these studies could be related to heterogeneous distribution of PE prevalence worldwide and with varying effectiveness of health interventions to reduce PE risk.[20,21] Another factor may be failure to quantify amount of alcohol, a limitation of Fang’s study as well as our own, in which alcohol use was treated as a nominal dichotomous variable.
It would be worthwhile to conduct further studies with amount consumed as a categorical variable quantified in a range from least to greatest values, to more accurately estimate associated risk; and to determine risk resulting from the interaction of this environmental factor, taking into account dose and genetic predisposition assessed by family history. It would also be useful to more accurately specify when alcohol intake took place, whether before or during pregnancy.
Multiple studies have demonstrated an association between age of the pregnant woman (<20 years and >35 years) and greater risk of PE. Authors in Thailand, Iran and Chile observed doubling of PE risk in such cases.[19,22,23] Suárez reported a high incidence of hypertensive disease of pregnancy in Cuban adolescents and women aged >35 years in Santa Clara during the period
January 2006–December 2008.[15,24] It is hypothesized that women aged >35 years have a greater frequency of chronic vascular diseases, leading to increased antiangiogenic factors and abnormal placentation, thus increasing susceptibility to PE.[25] It has been posited that abnormal placentation is more frequent in younger patients, consistent with the theory inadequate placentation as the etiological mechanism for PE.[25]
We consider age extremes at pregnancy to be modifiable risk factors, important for primary health care, since part of the primary prevention activities of the physician, family nurse, genetic counselor, and other professionals is prevention of pregnancy at extreme ages through education and timely preconception counseling. However, a multivariate logistic regression study is recommended to rule out possible confounding effects of variables such as smoking and age, which we did not do in this study.
Current thinking on PE pathogenesis is that it arises because of immune maladaptation between mother and conceptus, which can help explain why nulliparity is a risk factor.[26,27] During the first pregnancy, paternal antigens in the fetoplacental unit foreign to the host mother activate an immunological mechanism, posited as the trigger for the process leading to vascular damage, the direct cause of PE onset. In turn, immunological tolerance would also develop, which would prevent the disease from developing in subsequent pregnancies, provided the father was the same.[28,29]
Another possible explanation suggested for the association between nulliparity and PE risk is that because the nulliparous woman’s uterus has not previously been subject to distention of pregnancy, it has increased myometrial tone during the entire pregnancy, which reduces the caliber of the spiral arterioles by compression. This limits blood perfusion in the region, with the consequent possibility of trophoblast hypoxia, a phenomenon that has also been implicated in the physiopathology of PE.[4] These physiopathological underpinnings affirm nulliparity as an important risk factor, even though this study did not obtain this finding. This might be due to the small size of the study group and to the fact that we did not collect information on previous interrupted pregnancies in nulliparous women, both study limitations. It has been shown that a history of spontaneous or therapeutic abortion in primiparous women reduces PE risk.[30,31] We recommend that future research use a larger sample and include detailed inquiry into obstetric history, including previous interrupted pregnancies.
Smoking was statistically significantly more frequent in cases than in controls, suggesting a role as a risk factor for PE, as observed by Ioka.[32] However, some studies have described smoking as a protective factor, probably due to increased maternal placental growth factor and increased soluble fms-like tyrosine kinase-1 in maternal blood of women with abnormal uterine artery Doppler findings.[33] One limitation of our study was that smoking and tobacco use were treated as dichotomous variables; studies with both larger numbers and more detailed history about dose and duration of exposure could be more informative.
Analysis of the genetic component of PE Women whose first- and second-degree female relatives had PE were more likely to develop it. The increased risk was strongest if the affected relative was the grandmother. Interestingly, the husband’s female relatives’ PE experience also affected risk: a pregnant woman whose sister-in-law had PE had increased risk compared to pregnant women whose sisters-in-law had normal pregnancies.
The familial distribution of PE has been well established and is described by Chesley.[34] It is believed that first-degree relatives of affected women have two to five times greater risk of developing PE. In addition, there have been reports of risk of recurrence ranging widely, from 7.5% to 65%. With the development of molecular genetics, evidence has accumulated on the genetic contribution to PE susceptibility.[34–37] A study in Sweden demonstrated the existence of familial clustering in PE, finding ORs of 3.3 and 2.6 for sisters and daughters of a mother with PE,[38] consistent with our results.
Bezerra reported a statistically significant increase in PE risk in women whose mother or sister had the condition (p <0.007 and p <0.001 respectively). The association was stronger when both mother and sister had been affected (OR: 3.65).[39] Cruz found that female first-degree relatives of a woman who has had PE have a four to five times greater PE risk when they become pregnant. Similarly, second-degree relatives have a two to three times greater risk.[40] Berends also reported evidence of familial clustering in women with a history of PE, in a genetically isolated population.[41]
Chromosome regions associated with PE have been identified, but not yet specific genes. The most studied polymorphisms are: those related to vasoactive genes (M235T of the AT gene, I/D of the ACE gene, E298D of the eNOS gene); mutations of thrombophilic genes (1691G>A of Leiden factor V, 677C>T of the MTHFR gene, 20210G>A of the prothrombin gene); lipid metabolism and oxidative stress genes (Exon3 Tyr3His of the EPHX gene, Exon 6 Asn291Ser of the LPL gene); and genes involved in immune and inflammatory responses (-308G>A of the TNFα gene, -1082G>A of the IL10 gene); and genes involved in immunogenetics, placentation and genomic imprinting.[8,42,43] The additive action and interaction of some of these genes could explain the genetic predisposition for PE in some of the subject families in our study.
Heritability (h2) is the parameter that estimates the relative contribution of genetic factors to the appearance of diseases such as PE. In this study, it was greater than 0 but less than 0.75 (h2 = 0.24), which means that PE is not due exclusively to environmental factors, but must involve as well a genetic predisposition that is modulated by adverse environmental factors.
It should be noted that blood pressure values in pregnancy are a continuous variable due to the action of genetic and environmental factors or both, but the roles of these factors are still incompletely understood.
An interesting finding in this study is that PE risk increases when the husband has relatives with PE history, even when the woman’s own family history is negative. If the husband has a sister or mother with PE history, his wife’s likelihood of developing PE increases by factors of 6.71 and 1.99 respectively. Paternal genetic predisposition represented through the fetal genome is one more element in this network of interactions for PE occurrence.[44]
There is evidence in the scientific literature supporting fetal genetic effects on adverse pregnancy outcomes. Fetal genes from both parents are involved in control of placental and fetal tissue growth, and the placenta and membranes play an important role, which fetal genes could influence. Additionally, heritability studies estimating the relative portion of population variation in a genetically determined trait suggest that PE is influenced in part by fetal genetic factors.[45]
Fathers with a family history of PE increase the probability that their pregnant partner will develop it, but some authors report that a change in partners between pregnancies reduces risk of preterm birth in pre-eclamptic women.[46–49] These data suggest that paternal genes expressed in the fetus may contribute to PE. This has prompted studies of maternal–fetal influence, assessed by defining the infant as proband, rather than the mother, broadening the scope of investigation beyond specific maternal influences.[46–49]
The genome of the partner of the women in our study may present some of the molecular polymorphisms predisposing for PE previously mentioned, inherited from their probably pre-eclamptic mothers, and leaving a genetic imprint on the fetal genome.
Analysis of genome–environment interaction in PE Finally, the five statistically significant interactions that were found demonstrate how environmental factors tend to modulate genetic predisposition as reflected in family history. On a practical level, these findings could strengthen primary prevention through genetic counseling. Counselors would need to assess whether there is a clear probability of PE in the patient if she has a first-, second- or third-degree family history of PE (non-modifiable risk factor), since they have 50%, 25% and 12.5% genes in common respectively. However, there are modifiable environmental factors that could modulate this susceptibility.
In this study, we observed that coexistence of first-degree family history (such as an affected sister) with alcohol consumption increased PE risk by 16 times. If prevention activities had managed to eliminate alcohol consumption, this risk would have decreased the OR from 16 to 1.61.
The results tend to be similar for the rest of the interactions. It was shown that when environmental risk factors interact with presence of an affected first-degree relative (family history), probability of PE increased, more than if these factors were acting in isolation. ORs were always greater when a
non-genome-dependent predisposing factor interacted with the affected relative. Of these, alcohol consumption was the greatest.
It is interesting to note that there has been relatively little research on gene–environment and genome–environment interactions in PE. Since it is a multifactoral disease, genetic susceptibility to it will depend on other non-modifiable risk factors and modifiable ones (such as unhealthy behaviors) that could modify the expression of gene polymorphisms that predispose for PE.
The epigenetic regulation mechanisms described in the biology of the placenta are a focus of attention for researchers investigating environmental factors that modify expression of placental genes, and how these relate to diseases of gestation and the first years of life. Understanding epigenetic alterations in the placenta can inform diagnosis and prognosis of many diseases such as PE, and could help tailor health promotion and prevention activities.[50]
CONCLUSIONS
Our results, limitations discussed above notwithstanding, confirm that the risk attributable to the interaction of genetic and environmental factors is greater than the potential risk of these factors acting separately. This study found familial clustering of PE, representing the first Cuban evidence using genetic epidemiology family pedigree strategy addressing the following questions: Does PE preferentially cluster in families of cases vs. families of controls? Is this familial clustering due to genetic factors? A heritability coefficient of 0.24 demonstrates that both genetic and environmental factors influenced PE risk in the women studied.
These findings confirm the importance of preconception and prenatal care in primary health care, modifying environmental factors that could enhance or trigger polymorphic genes that predispose for PE. To this end, further studies of this type are needed in different regions of Cuba, building on the results presented here.
Finally, this first-of-its-kind Cuban study is important to maternal and fetal health. It is imperative to expand upon this initial effort by using multivariate analysis to develop Cuban tables for PE risk attributable to genetic predisposition, to adverse environmental factors and to their interaction.
This could support genetic counseling for PE in the community, taking advantage of the National Medical Genetics Network as a resource.[51] Such tables could also create more accurate risk perception by individuals and thus encourage health-promoting lifestyles, especially for otherwise healthy women with a genetic PE susceptibility.
Future studies of gene–gene and gene–environment interactions are imperative, as is delving further into possible epigenetic mechanisms, to characterize the most frequent gene polymorphisms in the Cuban gene pool.