INTRODUCTION
Cystic fibrosis (CF) is a multisystem disease affecting growth, respiratory function and digestion (mainly involving the lungs, intestines, sweat ducts and vas deferens). It presents wide variability in clinical severity[1] and is incurable. CF is characterized by elevated and premature mortality, as well as poor quality of life.[2] Symptoms vary, depending on mutation type, patient age, degree to which specific organs are affected, prior therapy and type of infection associated. The neonatal period is characterized by poor weight gain and intestinal obstruction. Other symptoms appear later in childhood and early adulthood, including growth retardation, lung disease and increasing problems related to nutrient malabsorption in the gut.[3,4]
CF is a health problem worldwide, constituting the most frequent autosomal recessive disease in European-descended populations.[5,6] It also presents a health problem in Cuba,[7] where, according to recent data from the National Cystic Fibrosis Commission, the disease occurs in 1 in 9862 live births. From 2001 through 2008, a total of 30 CF patients died, 33% of them aged <1 year.[8]
The gene responsible for CF was identified in 1989 and named the CF transmembrane conductance regulator gene (CFTR); the principal mutation, p.F508del, was also identified.[9] Since then, over 1900 mutations have been described.[10] Studies of various population groups have found considerable differences in frequencies of the most common mutations. Mutation p.F508del has been detected in 30%–90% of patients, depending on their ethnic origin.[11–13]
Given the ethnic origins of the Cuban population,[14] research began by detecting the most frequent mutations found in Spain’s population (p.F508del, p.G542X, p.R1162X, p.R334W and p.R553X) and the one most frequent in African populations (c.3120+1G>A). Only 55.5% of chromosomes with CF genes were identified; thus, Cuba’s population can be considered highly heterogeneous, making molecular diagnosis difficult.[15]
Direct diagnosis is the ideal method for detecting the genetic defect. This is quite feasible in populations without major molecular heterogeneity and with few mutations, but in very heterogeneous populations such as Cuba’s, it cannot be used in most couples potentially affected by CF. Indirect diagnosis is then indicated, using molecular markers such as XV2C and KM19 polymorphisms, and combinations of both methods—direct mutation detection and molecular marker analysis—to increase the chances of conclusive antenatal diagnosis.
Given CF’s severity, antenatal testing is important for at-risk couples, indicating to them whether their child may be healthy, affected or a carrier. This knowledge also facilitates tailored genetic counseling, as well as early treatment for better prognosis and quality of life for the baby, should the couple decide to continue with the pregnancy.
In Cuba’s public health system, such testing became available in the 1980s, first by enzyme analysis in amniotic fluid;[16] later by indirect analysis, using restriction fragment length polymorphisms (RFLPs) as molecular markers;[17] and finally, following cloning of the CFTR gene, by direct analysis of the gene’s mutations.[18] The country began antenatal molecular diagnosis in 1987 at the laboratory of the National Medical Genetics Center (CNGM, the Spanish acronym). Antenatal testing in Cuba is performed for all couples requesting it, whether carriers, CF patients, or couples for whom the disease is suspected on ultrasound. Cases are seen at provincial medical genetics centers and samples are sent to CNGM.
The objective of our research was to characterize the Cuban public health system’s experience with antenatal molecular testing for cystic fibrosis from 1988 through 2011.
METHODS
A retrospective descriptive study was done using CNGM laboratory data, results from testing on amniotic fluid from 108 fetuses of couples with some risk of having children affected by CF, referred from all parts of Cuba from 1988 through 2011.
Data collection Variables are described in Table 1.
Table 1: Variables
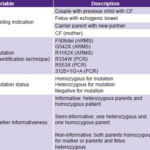
ARMS: amplification refractory mutation system / CF: cystic fibrosis
CNGM laboratory methods Amniotic fluid contaminated with maternal blood is discarded to prevent equivocal results. DNA is extracted by the salting-out method using 20 mL of amniotic fluid, as described by Miller.[19] Once DNA samples are obtained, they are coded and stored until use.
Markers XV2C/Taq I and KM19/Pst I are used for indirect analysis. They are detected by PCR and enzyme digestion by the respective restriction enzymes Taq I and Pst I, according to protocols described by Casals.[20] The process uses a thermal cycler (MJ Research, USA) and samples are analyzed by 2% agarose gel electrophoresis. Alleles are segregated for each marker in each case. When a child enters the program, parents are also tested for the markers of interest. Families are informative when both parents are heterozygous for the markers and the affected person is homozygous; semi-informative when one parent is heterozygous for the marker; and non-informative when both parents are homozygous for the marker, or parents and patient are heterozygous.
All sampes are tested for mutations p.F508del, p.G542X, p.R1162X, p.R334W, p.R553X and c.3120+1G>A.
Three mutations—p.F508del, p.G542X and p.R1162X(11)—are detected using the amplification refractory mutation system (ARMS).[21] The other three—p.R334W[22], p.R553X[23] and c.3120+1G>A—[24] are directly detected by enzyme digestion of the corresponding PCR product, amplified in a thermal cycler (MJ Research, USA) and fragments observed on 2% agarose gel stained with ethidium bromide.
Analysis Data were analyzed using summary measures for qualitative variables, absolute frequencies and percentages.
Ethical considerations The study was approved by the CNGM ethics committee and endorsed by the National Cystic Fibrosis Commission. Couples included had received genetic counseling and given written informed consent for antenatal molecular testing, including permission to use their data for future research. Patient privacy was protected throughout the analysis process.
RESULTS
Data were available for antenatal genetic testing conducted from 1988 through 2011, for 108 pregnancies of gestational age ranging from 16–20 weeks. Testing was repeated for four couples; one couple had a twin pregnancy.
Of the 108 antenatal tests carried out, 91 were for couples with a previous child affected by CF, the child having died in 43 (39.8%) of these cases; and 4 (3.7%) were carrier parents with a new partner. Fifteen couples (13.9%) were referred because ultrasound had detected echogenic bowel. In two couples (1.8%), one of the partners was a CF patient.
Case distribution for the 108 couples by antenatal diagnostic method was as follows: 93 (86.1%) diagnoses were made by direct analysis looking for mutations p.F508del, p.G542X, p.R1162X, p.R334W, p.R553X and c.3120+1G>A; 5 (4.6%) were diagnosed by indirect method using markers XV2C/Taq I and KM19/Pst I; and 10 (9.3%) combined the two methods.
Of the 93 antenatal tests searching for mutations p.F508del, p.G542X, p.R1162X, p.R334W, p.R553X and c.3120+1G>A, diagnosis was confirmed in 61 (65.6%) cases. Of these, there were 20 carriers, 13 fetuses with CF, and 20 healthy fetuses. Eight cases were healthy or carriers, who were free of the mutation detected in one parent, but had another unknown mutation, preventing confirmation of their genotype. Table 2 shows the genotypes of the 33 affected and carrier fetuses.
Table 2: Genotypes from antenatal CF testing

None of the cases referred due to echogenic bowel on ultrasound had a family history of CF, and all were negative for the tested mutations. Therefore, diagnosis could not be confirmed; and no later clinical diagnosis of CF was reported in any case.
One sign suspicious of CF is meconium ileus. One of the fetuses homozygous for the p.F508del mutation, which usually has a severe clinical presentation, had meconium ileus. There was no family history of CF.
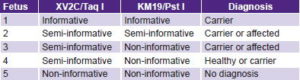
Five diagnoses were made using indirect study of markers XV2C/Taq I and KM19/Pst I. One family was informative; it was possible to identify which marker alleles segregated with the disease, confirming the fetus as a carrier. Three cases were semi-informative for one marker or the other; in one of the parents it was possible to identify which marker alleles segregated with the disease, finding two affected or carrier fetuses and one healthy or carrier fetus. The final case, homozygous for both markers, was non-informative (Table 3). Direct diagnosis could not be made in these cases because they were not carrying any of the six mutations detectable in the laboratory.
Table 3: Results of five antenatal CF tests with markers XV2C/Taq I and KM19/Pst I
Antenatal testing combining direct and indirect methods found four cases informative for the markers; in all cases, one parent was a carrier of the p.F508del mutation. Two fetuses were found to be carriers of p.F508del, and two were affected carriers of p.F508del and another unidentified mutation. The remaining six families were semi-informative for markers XV2C/Taq I and KM19/Pst I and one parent was a carrier of the p.F508del mutation. Of these, three fetuses were carriers of p.F508del, one was a carrier of an unknown mutation, one was affected (carrier of p.F508del and another unidentified mutation), and one fetus’s status could not be confirmed (Table 4).
Table 4: Antenatal CF testing with combination of direct and indirect methods

Conclusive diagnosis was possible in 72 cases (66.7% of those studied), of which 16 were affected, 27 carriers, 20 healthy, and 9 healthy or carrier fetuses.
DISCUSSION
Latin America is an extremely diverse region in population origin, culture and environment, CFTR gene mutation profiled across the region reflecting the heterogeneity of its inhabitants.[25] Cuba’s population today is the result of an admixture of contributions, mainly from Spanish and African ancestors.[26]
Great genetic heterogeneity in populations with a wide spectrum of CFTR gene mutations makes it difficult to detect mutations, limiting molecular diagnosis of the disease, especially in patients with uncharacterized mutations. Therefore, in Cuba, a combination of direct and indirect testing methods is needed to increase the chances for antenatal and patient CT-carrier diagnosis. Similarly, in the Mexican population, Chávez reports that the most frequent combination of molecular strategies (such as targeted search for CFTR mutations) with marker linkage analysis, is very useful for carrier detection and antenatal diagnosis in families affected by CF.[27]
Direct analysis for antenatal diagnosis of the CF molecular defect is only possible in families in which the specific mutation responsible for CF has been identified. In geographic regions such as Cuba, with great molecular heterogeneity and were the most frequent mutation (p.F508del) is less than 50%,[14] direct antenatal diagnosis is only possible in roughly half the cases. In this study, diagnosis could be confirmed with direct mutation detection in 65.6% of couples studied, which was expected due to the frequency of the F508del mutation and mutations p.G542X, p.R1162X, p.R334W, p.R553X and c.3120+1G>A reported in prior studies.[14]
Recently, other CFTR gene mutations have been identified in Cuban CF families, including I507del, G85E, 2184insA and G551D, also found in patients from other countries.[11] This will make direct mutation diagnosis possible for these families, although the low frequency of these mutations will not enable substantial overall diagnostic improvement, such as is possible in northern European populations in which p.F508del alone is present in roughly 70%–90% of cases.[28] Hence, indirect analysis will continue to be needed, in addition to direct mutation identification. Along with the RFLPs already in use, microsatellite markers[29] will be used, which are more informative than those utilized in the present study. In fact, this method has been standardized[30] and will be deployed in the future, increasing the number of couples that can be tested.
Although markers XV2C/Taq I and KM19/Pst I have been quite useful, they are not informative enough to sufficiently expand diagnostic possibilities, even though molecular diagnosis was possible in 40% of cases. One of the major limitations of indirect diagnosis in the families studied, in addition to lack of marker informativeness, was the death of the affected family member before molecular diagnosis could be done.
In Italy, a recent study of 181 couples at high risk for CF, testing failed to provide a diagnosis in a single case; of the remainder, 116 (64.4%) were diagnosed by mutation analysis; 40 (22.2%) required gene sequencing; 4 were tested for major rearrangements, and in 23, linkage analysis was done. In all, 42 of 180 (23.3%) fetuses were affected. The authors concluded that antenatal diagnosis of CF “should be performed in reference laboratories equipped for gene scanning and linkage analysis, with a multidisciplinary staff able to offer counseling to couples during all phases [of antenatal diagnosis].”[31]
Despite limitations preventing confirmation of all antenatal diagnoses in Cuban families affected by CF, this study was possible because molecular tools for antenatal testing are now universally available through the public health system (not the case in all Latin American countries), providing more accurate genetic counseling and enabling over half the families studied to learn their offspring’s status.
This facilitates more informed decisions by couples about the pregnancy, and if taken to term, about their child’s future: when CF is diagnosed, the infant can receive special care from birth, for better prognosis and quality of life.
All CF-affected families in Cuba continue to be offered molecular diagnosis, and the scope is increasing with the ability to detect more informative new mutations and molecular markers.
CONCLUSIONS
Implementation of direct and indirect testing methods enabled antenatal molecular diagnosis in over half the fetuses studied, which is of great social value because of CF’s burden on affected persons and their families.