INTRODUCTION
HIV-1 arose in the early 20th century from cross-species transmission of simian immunodeficiency virus from chimpanzees to humans in Central and West Africa.[1] HIV-1 group M, responsible for the majority of cases worldwide, initially spread throughout Africa and in response to various genetic forces, diversified into different subtypes, among them subtype B.[2–4] Spread of these variants in the human population was not noticed until 1981, when the first patients with symptoms of AIDS were described in the USA; they were infected with HIV-1 subtype B (HIV-1B).[5,6]
Spread of subtype B from Africa to the Americas initially occurred via its introduction into Haiti in the 1960s, probably with the return of Haitian professionals from work missions in the Democratic Republic of the Congo. Following this, it spread to the USA and from there to the rest of the world, as pandemic clade B.[7] Another subtype B lineage known as Caribbean B is found only in Haiti and neighboring Caribbean islands.[8] Because subtype B was the genetic variant isolated in industrialized nations and the first to disperse with population movements, its spread is the subject of continuing debate in the field of phylogeography.[9,10]
The first molecular epidemiology studies done in Cuba reported circulation of several genetic variants of HIV-1, with a predominance of subtype B in the HIV-positive population (primarily men who have sex with men).[11] Later studies also described high genetic variability, with multiple circulating recombinant forms, still with a predominance of subtype B.[12–18] Several studies by non-Cuban authors have analyzed the phylodynamics of Cuba’s HIV-1 epidemic, but have not taken into account the epidemiologic history of the patients studied.[19–21]
This study aimed to clarify the evolutionary history of HIV-1 subtype B in a sample of the HIV-positive population and to learn whether the subtype B virus variants transmitted are associated with variants detected at the epidemic’s outset. Another objective is to determine its phylodynamic pattern in the HIV-positive population several years after HAART use began in Cuba.
METHODS
Selection of HIV-1B nucleotide sequence group and multiple sequence alignment A retrospective descriptive study was done to confirm geographic relationship, time of most recent common ancestor (tMRCA) and evolutionary rate (µ, nucleotide substitutions per site per year) of Cuban subtype B sequences detected in resistance surveillance studies (120 sequences). We queried the database at the US Los Alamos National Laboratory on 29 January 2013 and retrieved data on 2021 sequences from protease (PR) and reverse transcriptase (RT) regions of the pol gene (nucleotides 2253–3233 in strain HXB2) of HIV-1B.[22]
To ensure high-quality data, sequences that met the following criteria[19] were selected:
- Cuban subtype B sequences reported in HIV antiretroviral resistance surveillance studies in 1999 (patients diagnosed in 1987–1999) and 2009–2012 (new diagnoses);
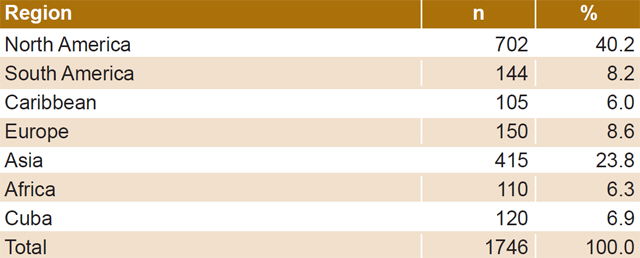
- Sequences representative of different geographic regions that include almost all continents (North America is overrepresented due to the greater availability of sequences) (Table 1);
- Sequences with no evidence of intersubtype recombination (The Recombinant Identification Program tool of the US Los Alamos database was used,[22] with a minimum confidence interval for pure subtype B of 0.95 and a window size of 200 nucleotides); and
- Sequences with no evidence of hypermutations, stop codons or excess of undetermined nucleotides.
These criteria were used to select 1746 sequences for analysis, which included 120 Cuban subtype B sequences studied in 1999 and 2009–2012.
Table 1: HIV-1B pol gene sequences by geographic region

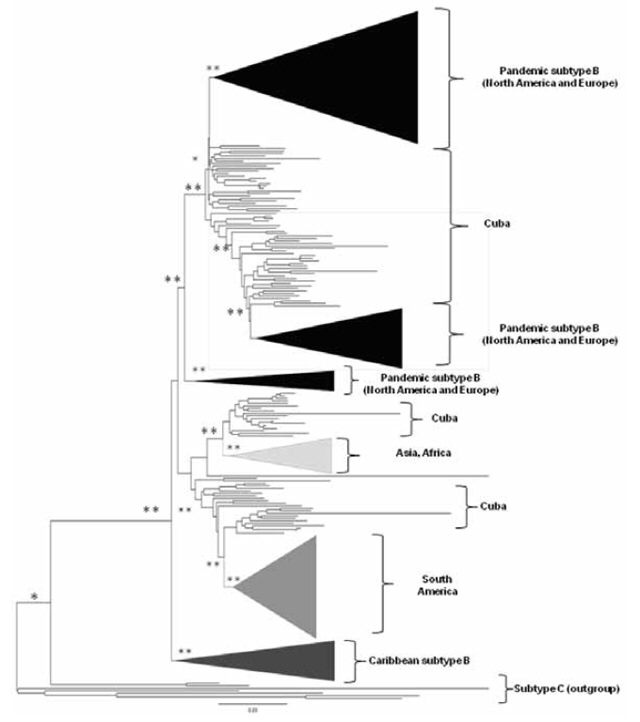
Figure 1: Phylogenetic tree of 1746 sequences of the HIV-1B pol gene

*bootstrap value >0.80 **bootstrap value >0.90 Tree constructed using neighbor-joining; genetic distance estimated by calculating matrices according to Kimura’s two-parameter method. Sequences from other geographic regions were grouped and are represented with different tones.
Sequences were aligned using the Clustal X tool of Bioedit Sequence Alignment Editor[23] and were manually edited. Two subtype C sequences were used as outgroups.
Phylogenetic analysis and reconstruction of evolutionary and demographic history To determine the geographic relationship of Cuban HIV-1B sequences to other strains from other regions, several phylogenetic trees were constructed using genetic distance, maximum likelihood and Bayesian inference methods. The tree with the greatest reproducibility and consistency was chosen, which ended up being the one constructed with the neighbor-joining similarity method and estimation of genetic distance based on Kimura’s two-parameter method.[24] Bootstrap values were calculated using 1000 replicates, with MEGA v 5.0.[25]
A BEAST program input file was generated using the BEAUti tool from BEAST v. 1.7.4.[26] Evolutionary rate and tMRCA were estimated according to the best demographic model. The best coalescent model was determined by estimating log likelihood with the Akaike information criterion through Markov chain Monte Carlo (AICM). Several parametric coalescent demographic models were used (constant population size and exponential growth) and a nonparametric one, the Bayesian skyline plot, which were compared under strict and relaxed molecular clock conditions, with uncorrelated lognormal distribution (UCLD), and with uncorrelated exponential distribution (UCED).
The best coalescent model was chosen by applying the Bayes factor through log likelihood. Markov chain Monte Carlo was run for 300 million generations to obtain an effective sampling size of >250 (the value for which the method is considered reliable) for the parameters of interest. As recommended in the BEAST program manual, 10% of trees constructed were discarded.[26] Convergence was measured via estimation of effective sampling size after constructing 10% of the trees using Tracer v. 1.5.[26] The trees were plotted using FigTree v. 1.1.2.[27]
Ethics Ethical procedures met the standards of the Cuban Ministry of Public Health and of the Ministry of Science, Technology and the Environment, in conformity with the principles of the Helsinki Declaration.[28] The study was reviewed and approved by the Bioethics Committee of the AIDS Research Laboratory and included the majority of recently diagnosed adults reported in surveillance of transmitted HIV-1 resistance to antiretroviral drugs in Cuba. Data management procedures ensured patient confidentiality.
RESULTS
Geographic relationship, origin and evolutionary rate of Cuban HIV-1B sequences The majority of Cuban HIV-1B sequences were grouped with viruses from North America, as shown on the phylogenetic tree (Figure 1). Other sequences were related to viral variants from other geographic regions.
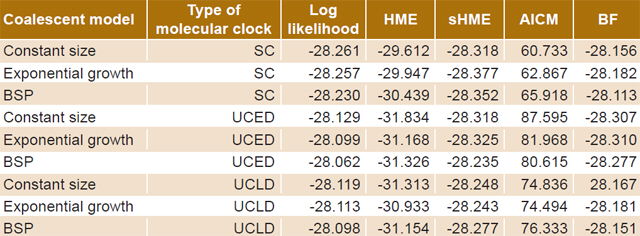
Table 2 displays the values obtained for the different coalescent models. The model chosen for analysis was the Bayesian skyline plot with a UCED strict molecular clock, because it showed the highest log-likelihood values.
Table 2: Demographic models of HIV-1B evolution

AICM: Akaike information criterion through Markov chain Monte Carlo BF: Bayes factor BSP: Bayesian skyline plot HME: harmonic mean estimator SC: strict clock sHME: smoothed harmonic mean estimator UCED: uncorrelated exponential distribution UCLD: uncorrelated lognormal distribution
The analysis estimated 1977 as the tMRCA date for Cuban sequences (95% CI 1974–1982) and estimated µ to be 2.73 x 10-3 substitutions per site per year (95% CI 2.37–3.09 x 10-3).
DISCUSSION
The HIV-1 epidemic in Cuba is characterized by elevated genetic diversity, which manifests in widespread circulation of subtypes and multiple recombinant forms.[11–18]
Although comparisons among countries and population groups and across studies are difficult, due to differences in the gene being analyzed, the methods used to align multiple sequences, the nucleotide substitution models and the degree of homogeneity of data used in the estimation,[29] the grouping of Cuban sequences with sequences of diverse geographic origins, primarily from North America and Europe, suggests that this subtype was introduced into the country through multiple events. This result concurs with early epidemiologic studies that detected subtype B in Cuba, and reported the infection source to be the USA and other countries, such as Ethiopia, Angola, the Democratic Republic of the Congo and Spain.[11] Furthermore, presence of independent groupings of Cuban sequences indicates indigenous transmission of this viral variant in the population diagnosed with HIV-1 infection during 2009–2012. In Cuba, subtype B had its founder effect in populations of men who have sex with men, facilitating its rapid dissemination[30] and making it the most common variant reported in the HIV-1 database of the US Los Alamos National Laboratory (34.4%).[22]
Delatorre and Bello, who employed all Cuban sequences of the HIV-1 pol gene from 2001 to 2012 available in the Genbank database, stated that the bulk of subtype B infections in Cuba resulted from dissemination of a few founder viruses introduced from North America and Europe.[21] However, another study that analyzed the spread of this variant in the Western Hemisphere employed only 12 Cuban sequences obtained in 1999 by Pérez[14] and associated the origin of Cuban subtype B with viral strains from South America.[19]
Neither group of authors took into account the epidemiologic history of HIV-1 in Cuba; they relied on computational methods, mostly based on biological samples obtained after the estimated time of infection, which varies from case to case.[31] With respect to origin and dissemination of subtype B in the Western Hemisphere, Junqueira posed two epidemiologic scenarios: direct introduction of subtype B to South America, with a later secondary outbreak in the USA, and spread of subtype B from the Caribbean to North and South America.[19]
Spread of subtype B from Africa to the Americas initially occurred through the introduction of this subtype into Haiti in the 1960s, probably associated with the return of Haitian professionals from work missions in the Congo.[3,4,7,10] However, it must be taken into account that direct introduction of subtype B into the USA from the Caribbean generated a genetic fingerprint in the viral genome that is evident when studying appropriate genetic markers, such as the pol gene, which is why the phylogenetic patterns expected under such models could group a significant number of US sequences early in the epidemic with Caribbean strains.[19] However, phylogeographic reconstruction by Pagán and Holguín in 2013 indicated that subtype B in the Caribbean could have originated in Puerto Rico and Antigua, and that this genetic variant in Puerto Rico, in turn, possibly originated in the USA.[9,20]
Our study estimated that HIV-1B appeared in Cuba in the late 1970s. This result falls within the confidence interval of tMRCA obtained by Pagán and Holguín in 2013, when they reconstructed the time and routes of dispersion of the subtype B epidemic in the Caribbean and Central America and estimated 1982 as tMRCA for Cuban subtype B (95% CI 1975–1985).[20]
In contrast, other authors place tMRCA for Cuban subtype B sequences at 1991 (95% CI 1988–1994) and 1992 (95% CI 1986–1994).[21] When analyzing viral sequences from Haiti that grouped with African sequences, Gilbert posed the hypothesis that subtype B spread in 1966 from Africa to the Americas (95% CI 1962–1970) and that Haiti was the epicenter of the introduction of subtype B into the USA before its global dissemination.[32] Keeping in mind that the first AIDS cases were described in patients in the USA in 1981,[33] there is a relationship between the estimated tMRCA obtained by Junqueira[7] and the natural history of the disease, given that there is a difference of 15 years from its possible origin and progression to AIDS; 5%–10% of patients remain asymptomatic from 10–15 years following infection and maintain a CD4+ lymphocyte count of >500 cells/mm3 (patients with nonprogressive disease).[34]
In Cuba, the first cases of HIV infection were diagnosed in 1986, and the geographic origin of several viral variants was determined epidemiologically. In 1995, Rolo analyzed 58 sequences from the C2–V3 region of the HIV-1 env gene, from Cuban patients infected by HIV-1 in the USA, Ethiopia, Angola, Spain, Democratic Republic of the Congo, and Cuba. Some 41 of these sequences corresponded to subtype B, with year of diagnosis ranging from 1986 to 1993.[11] In a later study to assess the emergence of antiretroviral drug resistance, pol gene sequences in 106 samples from Cuban patients diagnosed with HIV-1 infection from 1987 to 1999 indicated that 48.1% of samples corresponded to subtype B.[35] Our results for tMRCA are consistent with the epidemiologic history of the Cuban epidemic to date.
Delatorre and Bello estimated tMRCA at 1991 for Cuban subtype B, much later than the estimated origin of subtype B in Haiti and the USA (1960–1970).[21] Their explanations are based on the fact that the tMRCA they obtained for Cuban subtype B coincides with the economic crisis caused by the disintegration of the Soviet bloc in 1991 and the increase in tourism from North America and Europe, regions with extensive circulation of pandemic subtype B.[21] Several elements lead us to disagree with their conclusions.
First, in selecting sequences to study, Delatorre and Bello excluded those with mutations associated with antiretroviral resistance,[21] an aspect that according to several authors does not affect phylogenetic reconstruction of HIV-1, and that enables a more accurate estimate of the evolution of the infection.[36,37] Second, in selecting the best coalescent model, they ran Markov chain Monte Carlo for only 50 million generations,[21] which might interfere with obtaining an effective sampling size of >250, the value considered reliable for analysis.[27] Finally, they did not consider the epidemiologic history of the Cuban epidemic and overlooked the first studies that described circulation of subtype B in Cuba based on samples from patients diagnosed with HIV-1 infection as early as 1986.[11,35]
Estimated evolutionary rates indicate a rapid dynamic in HIV-1B viral populations. These figures may be overestimated due to the short time frames and presence of mutations that can be a common in intraspecific analysis of evolutionary rates.[38] The evolutionary rate obtained in our study is within the range described for other HIV-1B populations in the world.[20,21,39–42] This high evolutionary rate may be due to the fact that the region of the pol gene that codes for PR and RT enzymes is subject to selective forces, unsurprisingly, since these proteins are involved in functions in the viral replication cycle, such as infectivity and replication.[43] However, Pagán and Holguín also described a high degree of genetic conservation in the pol gene, which contradicts the high evolutionary rate found.[20] This coexistence may be the result of multiple mutations in a small number of sites located on the HIV-1 pol gene,[44] and the fact that, to be functional, these proteins require complete preservation of at least two-thirds of their sequences.[45,46]
CONCLUSIONS
Our results suggest multiple introductions of HIV-1B into Cuba in the late 1970s, predominantly strains from North America and Europe. These results underline the importance of maintaining, reviewing and updating the molecular epidemiology of HIV-1 in Cuba, due to the virus’s rapid evolution and implications for the Cuban Ministry of Health’s National STI/HIV/AIDS Program.