In monocytes, basal cytokine levels are elevated, antigen-presenting molecules are decreased, and phagocytosis changes.[35] In dendritic cells, receptor expression, antigen capture, migratory capacity, functions related to motility, the capacity to activate T cells, and the secretion of cytokines with a decrease in IL-12 and an increase in IL-23 are all affected, while myeloid dendritic cells remain in a semi-activated state that can lead to cytokine secretion even at baseline.[65]
Neutrophils are at the high end of normal limits. Their chemotaxis, respiratory burst resulting in free radical production,[64] microbial functions, phagocytic activity and degranulation capacities are all affected.[66] Neutrophil migration patterns are affected and neutrophil proteinase release occurs, contributing to tissue damage and systemic inflammation.[66] Natural killer (NK) and mature NK cells increase in number,[62] but cytokine secretion decreases[64] (mainly IFN-γ)[67] as does perforin production.[66] Although lytic activity is reduced,[62] its overall activity appears to remain intact and stable.[67]
Changes in the balance of T lymphocyte populations are the result of thymic involution. As part of this alteration, the number of naïve T lymphocytes decreases, memory T lymphocytes accumulate[58] and activated effector cells are dysfunctional and less diverse.[58,63] There is a reduced response to stimulation, and damage to proliferative response with decreased TCD4+ lymphocytes.[62] Functional alterations in Th1 and Th2 lymphocytes produce changes in cytokine secretion with decreases in IL-2 and increases in IL-4 and IL-5.[62] There is also a decrease in naïve TCD8+ lymphocytes and an increase in senescent-memory TCD8+ cells,[68] and therefore a reduction in T cell reserves necessary for protecting the host against novel pathogens.[58,61,69]
Primary antibody responses are weak and short-lived,[67] with lower antibody specificity, affinity and isotype switching.[59] Aging bone marrow has a reduced ability to fully ensure hematopoietic regeneration,[67] which results in peripheral virgin-cell depletion.[63] The spleen suffers a loss of germinal centers and reduction in frequency and size of plasmacytic foci.[67] This results in a decrease in specific antibodies, favoring short-term maintenance of total immunoglobulin levels and serum antibody titers at the expense of ability to establish fully functional memory B cell levels.[67] All of this leads to increased levels of pro-inflammatory markers and accumulation of activated dysfunctional effector cells with limited repertoires,[48,59] resulting in age-related inflammation.[28] This is a physiologic response to lifelong antigenic stress, altered mitochondrial function, accumulated oxidative stress and compromised antioxidant defense systems.[28,46]
Chronic immune activation, generated in part by persistent antigenic stimulation, contributes to an inflammatory state of multifactorial origin, leading to telomeric erosion and replicative senescence. Replicative senescence, in turn, results in the accumulation of senescent and exhausted T lymphocytes.[28,59] Dysfunctional or hypofunctional cells with terminally differentiated phenotypes express inhibitory molecules such as PD-1 and cytotoxic lymphocyte-associated protein 4 (CTLA-4).[28,58] This depleted phenotype can be also be induced by chronic infections in younger individuals,[70] and can be reversed, at least partially, by therapeutically blocking the immune system’s so-called ‘checkpoints’ (PD-1, CTLA-4, lymphocyte-activation gene 3 or LAG-3, T cell immunoglobulin and mucin domain 3 or TIM-3 and TIMIN).[28]
This knowledge has made it possible to recognize the modifiable nature of some factors involved in immunosenescence. Immunosenescence can be viewed as the result of continuous antigenic challenges[70] induced by inflammatory aging and vice versa,[28] thus understood as two sides of the same coin.[60] The pro-inflammatory state typical of aging is characterized by the accumulation of senescent cells and massive secretion of molecules producing the aptly-named senescence-associated secretory phenotype (SASP).[36] SASP cells produce cytokines, growth factors and proteases that influence neighboring cells, converting them to newly senescent cells through the so-called ‘bystander effect.’[56] However, exceptionally successful centenarians are equipped with well-preserved and efficient immune mechanisms, likely through optimal combinations of lifestyle and fortuitous genetics.[61,71]
Healthy centenarians have immune functions similar to those in the young, suggesting that long-lived individuals have less immunosenescence.[70] Keeping chronic low-grade inflammation under tight control is a driving force for longevity due to counter-regulation by anti-inflammatory molecules.[28] Centenarians’ survival is associated with immune systems that no longer react strongly to persistent infections.[58]
Centenarians possess trained immune system memories regulated by epigenetic changes, the key to controlling the inflammation characteristic of aging.[28] Their peripheral blood mononuclear cells respond well to chemotactic stimuli, and there is an increase in highly active NK (CD16+CD57-) cells with well-preserved cytotoxicity.[61,71] T lymphocytes show an inverted immune risk profile (a high CD4/CD8 ratio and a lower number of CD8+CD282 cells).[50] There is very little autoimmune response, a marked decrease in B lymphocytes,[61] and an almost complete lack of organ-specific autoantibodies.[61,71] Centenarians also have a well-conserved number of T lymphocytes capable of proliferating, but with delayed maximal responsiveness.[61] This delay could constitute a possible risk factor for more severe manifestations of COVID-19, to which we should also add the deterioration of the immune barrier systems that play an important role in natural resistance to infection.
Chronic noncommunicable diseases Since the beginning of the COVID-19 pandemic, many studies have examined the association between its unfavorable clinical evolution and chronic diseases like metabolic disorders, cardiovascular disorders, oncoproliferative diseases and chronic respiratory illnesses, among others.
A meta-analysis of 24 articles including 10,948 COVID-19 patients found that the presence of chronic disease is related to greater COVID-19 severity (OR 3.50) and greater likelihood of intensive care unit (ICU) admission (OR 3.36). The main comorbidities influencing this increase in severity are diabetes mellitus, hypertension, COPD and cardiovascular disease (OR 2.61, 2.84, 3.83 and 4.18, respectively).[14] Another meta-analysis of 43 studies with 3600 patients reported similar results regarding median prevalences of hypertension (16.0%), diabetes (10.1%) and COPD (2.0%), but did not relate it to the probability of developing severe forms of COVID-19.[15] Yet another meta-analysis of 19 studies involving 656 COVID-19 patients concluded that the disease is accompanied by high morbidity in chronic disease patients (36.8% of patients), among which the most common were hypertension (18.6%), cardiovascular disease (14.4%) and diabetes (11.9%).[16] These data coincide with a study that found that of the total number of COVID-19 cases admitted to ICU, 72.2% had comorbidities, contrasted with only 27.5% of patients who did not require ICU admission.[17]
Jain’s meta-analysis, reviewing 7 studies involving a total of 1813 COVID-19 patients, found that despite its rarity, COPD carries a high risk for progression to severity (OR 6.42, 95% CI 2.44–16.9) and the need for ICU admission (OR 17.8, 95% CI 6.56–48.2) and is the comorbidity with the highest predictive capacity for these events.[18] A meta-analysis including 1558 COVID-19 patients in 6 studies[72] also found an association between COPD and poor patient progression (OR 5.97). This association is largely due to changes in innate pulmonary defenses, namely those of immune barriers, that characterize COPD.
Total number of COVID-19 deaths is associated with obesity.[73] In a study by Muscogiuri,[74] obesity and morbid obesity were present in 47.6% and 28.2%, respectively, of patients requiring mechanical ventilation. A meta-analysis by Kumar,[75] including 33 articles and 16,003 patients, showed that diabetics who contract COVID-19 are 2.7 times more likely to develop severe infection and dying than non-diabetics, and that this risk increases with age. Obese or diabetic patients are particularly vulnerable, and the incidence of diabetes mellitus is twice as high in COVID-19 ICU patients than in those who do not require intensive care.[75]
Hu’s meta-analysis of 21 studies and 47,344 COVID-19 patients[76] reported a cancer incidence of 1.2%. A similar proportion was reported by El Gohary´s meta-analysis of 22 studies, which found 2.1% of cancer patients among the 11,243 COVID-19 patients involved, of whom 45.4% had serious or critical manifestations of the disease and 21.1% died.[77] In Wang’s meta-analysis of 138 hospitalized COVID-19 patients, 7.2% had cancer, 40% of whom required ICU admission, representing 11.1% of all 138 cases admitted to the ICU.[17] Liang’s study of 1590 COVID-19 patients found that cancer patients infected with SARS-CoV-2 have a 3.5 times higher risk of serious events, including ICU admission or invasive ventilation, than patients without cancer, among whom age ≥60 years is an aggravating factor.[78] Another analysis (of 6 studies and a total of 1558 patients) failed to find any statistical significance between cancer and severe COVID-19 manifestations (p = 0.49), despite an OR of 2.3.[72] This variability may be due to differences in neoplasm location, stage, evolution and treatment, which was not taken into account in these analyses.
Metabolic disorders Inflammation stimulated by metabolism, called metainflammation, is an important aspect of metabolic disorders[79] such as obesity, insulin resistance, type 2 diabetes and fatty liver disease.[46] Metabolic imbalances in these diseases coincide with the cellular processes involved in premature aging, suggesting the two may be related.[79]
Obesity Now a global epidemic, obesity involves genetic, epigenetic, hormonal and lifestyle factors,[80] and contributes to development of other metabolic diseases.[74] It is a serious health problem associated with cardiovascular diseases and certain types of cancer,[81] and is accompanied by chronic subclinical inflammatory processes that can affect immune response to infection through direct, indirect and epigenetic mechanisms.[82]
The role adipose tissue physiology plays in the immune system has been understudied.[81] Adipose tissue functions as an endocrine organ that secretes adipokines, growth factors and cytokines,[80] which act in an autocrine or paracrine manner,[46] regulate various metabolic processes[80,81] and modulate inflammation.[48,80] It contains mesenchymal stem cells, stem cells derived from adipocytes, endothelial cells and fibroblasts, which, among others, are responsible for producing extracellular matrices and spontaneous cellular regeneration or induced cell turnover under cellular stress.[79]
Obesity alters the composition, structure and function of adipose tissue,[79] and produces dysfunctional tissue[40,80] whose expansion is accompanied by inflammatory changes that contribute to systemic low-grade inflammation,[80] with inflammatory cell infiltration and endoplasmic reticulum stress production, mitochondrial dysfunction, hypoxia, fibrosis, cell senescence and changes in lipid metabolism.[81] Adipose tissue inflammation initiates systemic inflammation in central (visceral) adiposity.[46] This condition reduces immune cell functionality, results in an imbalance of the intestinal microbiome/virome and induces an inflammatory cytokine phenotype.[82] Macrophages are the main cell type that infiltrate expanding adipose tissue in response to chemokines (MCP-1) produced by hypertrophic adipocytes[80] that increase in number and change location, phenotype and inflammatory characteristics under conditions of obesity.[83] Monocytes, on the other hand, connect low-grade chronic inflammation and altered lipid metabolism through the expression of inflammatory mediators and by modulating intracellular lipid accumulation.[84]
Additionally, TCD4+ and TCD8+ lymphocytes increase, Treg lymphocytes decrease, and this induces a phenotypic shift, recruiting Th1 lymphocytes from the periphery to aid in remodeling the extracellular matrix.[80] This process is associated with fibrosis, insulin resistance and metabolic dysfunction.[79] Th1 lymphocytes secrete interferon, which stimulates production of various chemokines that extend T lymphocyte migration into adipose tissue, including chemokine C-C motif ligand 2 (CCL2), chemokine C-C motif ligand 5 (CCL5), chemokine C-X-C motif ligand 9 (CXCL9) and chemokine C-X-C motif ligand 10 (CXCL10).
Body fat mass increase in children is associated with development of an early immunosenescent profile. Obese children have lower shares of naïve TCD4+ and CD8+ lymphocytes, higher shares of effector memory lymphocytes, and more intermediate TCD8+ lymphocytes and late and senescent TCD4+ lymphocytes; all characteristic of an immunosenescent profile. Obese children also show decreased response to vaccines and increased susceptibility to infection, also characteristic of immunosenescence.[79] Senescence itself impairs normal adipose tissue function and probably contributes directly to the inflammatory characteristics of adipose tissue in obese individuals.[81]
The number of B lymphocytes also increases with body mass index (BMI).[48] The increase in pro-inflammatory B cells is the main driver of the inflammatory profile of T lymphocytes seen in obesity and type 2 diabetes. These cells are characterized by increases in basal secretions of IL-6 and IFN-γ, accompanied by marked decreases in IL-10 secretions, a combination that results in chronic low-grade inflammation.[80] Cytokines released by B cells contribute to phenotypic changes in adipocytes in the abdomen, causing them to release adipokines, other pro-inflammatory markers and cellular debris.[49]
Obese COVID-19 patients experience an interaction between the immune system and adipose tissue, which favors immune system attenuation and chronic inflammation.[82] Proposed mechanisms for the increased severity of the disease in obese patients include reduced end-expiratory volumes, positive end-exhalation pleural pressure, low-grade chronic inflammation and altered immune responses to infection.[53] Barrier mechanisms can become compromised by hypoventilation syndrome, obstructive sleep apnea and other mechanical abnormalities caused by excess thoracic and abdominal fatty mass, leading to respiratory complications.[74] Other factors present in obese patients that potentiate severe forms of COVID-19 are an increase in clearly pro-inflammatory Th17 lymphocytes,[82] vitamin D deficiency,[73,85] and intestinal dysbiosis,[74,85] which can also impact respiratory system mucosa, affecting their function as protective barriers.[28,48.74] These last two risk factors are modifiable and possible points of preventive intervention, including vitamin D therapeutic supplements and consumption of prebiotics and probiotics that can effectively restore healthy gut microbiota. However, as these interventions require long treatment periods before benefits are accrued, they would have little impact during the course of a SARS-CoV-2 infection.
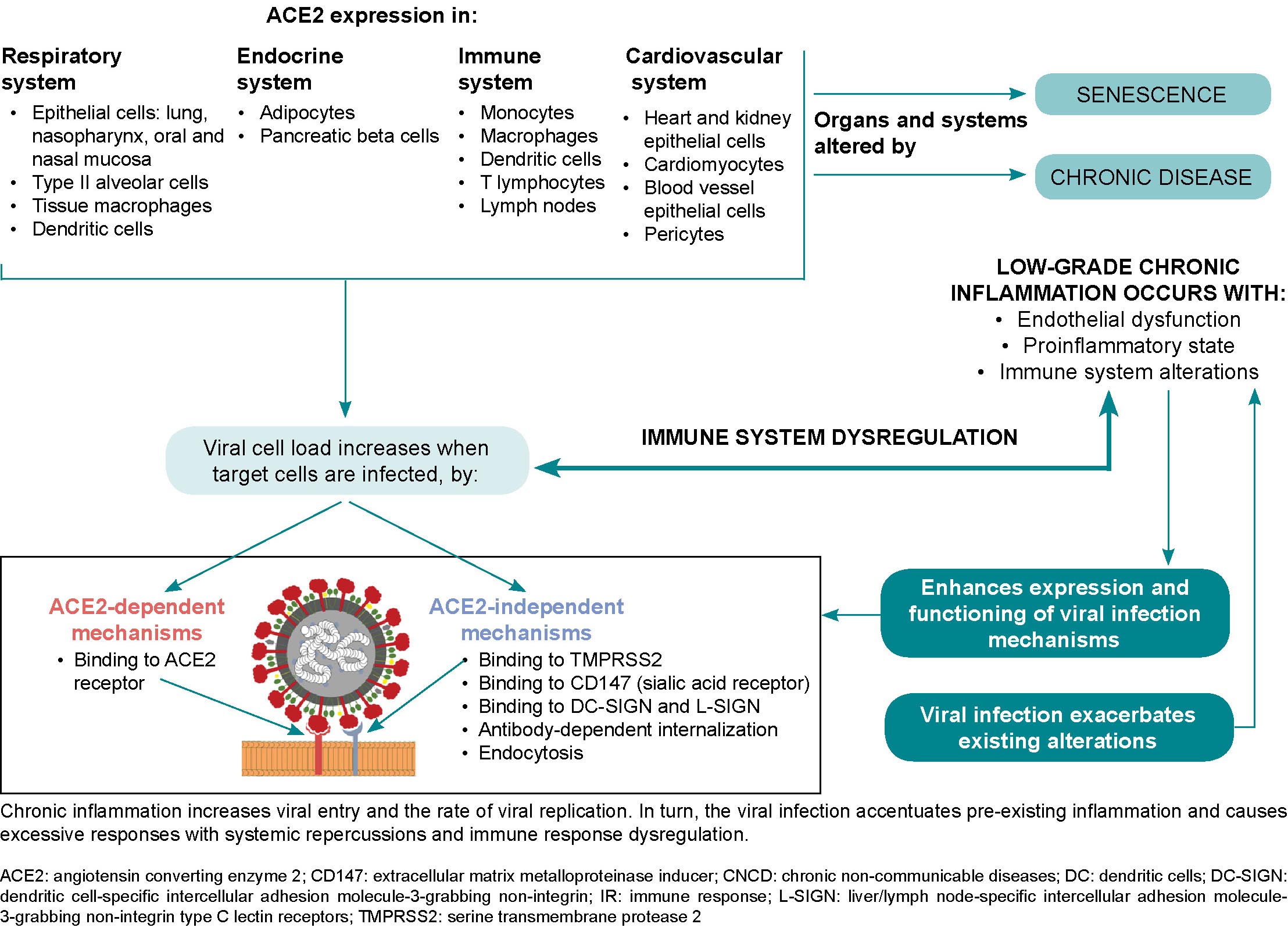
SARS-CoV-2 can infect subcutaneous and visceral adipose tissue, since angiotensin-converting enzyme 2 (ACE2) expression levels are higher in these tissues than in lung tissue.[86,87] Additionally, in obese patients, angiotensin-converting enzyme 1 (ACE1) increases, ACE2 is inhibited, and angiotensin II activates angiotensin I and II receptors, mediating a pro-inflammatory response and a consequent increase in vascular permeability.[82] Adipose tissue may act as a reservoir for the virus and may contribute to viral shedding, increased immune activation and cytokine amplification, explaining obese COVID-19 patients’ poor prognoses.[73]
Type 2 diabetes mellitus There has been a parallel increase in the incidences of obesity and diabetes, to the point that diabetes is often considered a comorbidity of obesity.[80] Individuals with this type of diabetes experience both inflammation and metainflammation, most frequently seen in older adults in whom immune disorders overlap with immunosenescence. Type 2 diabetes driven by inflammation is observed at an early age, suggesting that it is a model of premature immunosenescence, as senescent or late-differentiated T lymphocytes predict development of hyperglycemia in humans. Additionally, a feature common to both diabetes and old age is a decrease in naïve TCD4+ lymphocytes, with an increase in memory TCD4+ lymphocytes and effector TCD4+ and TCD8+ lymphocytes.[79]
The immune system is compromised in diabetic patients, especially innate immunity,[88] with phagocytic cell dysfunction and neutrophil chemotaxis inhibition, leading to an uncontrolled inflammatory response, increased levels of enzymes related to tissue damage and states of hypercoagulation, significantly elevated serum levels of inflammatory biomarkers and a proinflammatory cytokine profile.[74] Transient hyperglycemia may temporarily affect the innate immune system’s response to infection,[88] as it serves as a trigger for proinflammatory cytokines in innate immune cells, particularly IL-1, IL-6 and TNFα.[79] One mechanism of immune impairment driven by hyperglycemia is the overactivation of the advanced glycosylation end-product pathway and its receptors. Additionally, elevated levels of glycated hemoglobin are associated with decreased phagocytic activity in circulating monocytes and neutrophils.[79] Therefore, maintaining optimal glycemic control in COVID-19–positive diabetics can help to prevent complications and more severe manifestations of the disease.
Factors contributing to disease severity in COVID-19–positive diabetics include hyperglycemia, impaired immune function, suboptimal glycemic control during hospitalization, prothrombic and proinflammatory states,[53,88] and reduced forced vital capacity (FVC) and forced expiratory volume in one second (FEV1) in pulmonary function tests.[88] Fatty liver disease frequency in type 2 diabetics may increase the risk of an exaggerated immune response, including development of a cytokine storm, which is associated with severe lung injury in COVID-19 patients.[53]
Current evidence does not support the conclusion that diabetics are more susceptible to developing SARS-CoV-2 infection, but they are more likely to have more severe clinical manifestations.[88] In turn, COVID-19 can induce diabetes by either increasing insulin resistance or by direct damage to the islets of Langerhans,[38] which have abundant viral ACE2 receptors (detectable by immunostaining),[69] whose damage leads to reduced insulin release.[88] ACE2 and dipeptidyl peptidase-4 (DPP4), in addition to acting as receptor proteins for the SARS-CoV-2 virus, are also metabolic signal transducers that participate in pathways that regulate inflammation, renal and cardiovascular physiology and glucose homeostasis.[88] SARS-CoV-2 infection can cause sharp fluctuations in blood glucose levels, and glucose metabolism dysregulation both exacerbates diabetes and increases infection severity.[88] The resulting proinflammatory environment and possible cytokine storm exacerbate existing inflammation and result in systemic inflammation with serious consequences,[74] compounding damage already present in diabetic patients.[38]
Cardiovascular disease Hypertension is one of the most common diseases worldwide, and is considered a silent killer.[89] It is the most common modifiable risk factor in cardiovascular disease,[90] frequently coexisting with other comorbidities and with no apparent cause in 90% of cases.[91]
The immune system plays an important role in hypertension[92] via low-grade inflammation in the kidneys, the arterial wall, the central nervous system,[93] the sympathetic nervous system[91] and the heart,[92] which favor both development and increased severity of hypertension.[93] Low-grade inflammation has been considered both a cause and a consequence of hypertension.[90] In chronic inflammation, innate and adaptive immune cells are activated, cause organ and tissue damage through production of various cytokines and chemokines,[91] contributing to further immune system dysfunction, blood pressure elevation and vascular remodeling.[92]
Other changes in hypertensive individuals include increased pattern recognition receptors; NK cell migration toward the aortic wall; increased serum levels of complement proteins C3a and C5a;[90] increased cyclic dilation of larger vessels; release of IL-6, IL-8, endothelin and other proinflammatory mediators; and increased endothelial expression of VCAM-1, ICAM-1 and cluster of differentiation 40 (CD40).[91] These changes enhance activation of adjacent monocytes, macrophages and dendritic cells. T cell receptors, adregenic receptors and mineralocorticoid receptors on CD8+ cells play important roles in promoting IFN-γ production.[91] The TCD4+ immune response is polarized toward Th1 and Th17 phenotypes, IL-17 secretors and other cytokines involved in hypertensive inflammatory mechanisms.[90]
T lymphocyte polarization—which also occurs in antiviral response—can elicit exaggerated immune responses in hypertensive patients. Poor blood pressure control leads to increased dysregulation of the immune system.[42] Hypertension results in increased blood levels of monocytes, eosinophils and neutrophils.[92] A relationship has been suggested between increased lymphocyte levels and hypertension.[92] CD8+ T lymphocyte dysfunction caused by increased immunosenescent, proinflammatory and cytotoxic CD8+ T lymphocytes has been observed in hypertensive patients.[83,94] These lymphocytes cannot effectively fight viral infections,[42] and increase production of IFN-γ, TNF-α and the cytotoxic molecules granzyme B and perforin,[91,94] contributing to the pathological overproduction of cytokines that occurs in low-grade inflammation.[42]
Animal models suggest that lymphocytic effects may also be modulated by vascular function, blood flow and sympathetic nervous system activation. Endothelial dysfunction favors the recruitment and activation of monocytes and antigen-presenting cells that facilitate inflammation through the release of pro-inflammatory cytokines, which increases endothelial dysfunction, closing a vicious circle in hypertension pathogenesis.[92] Effective antihypertensive treatment is believed to help restore dysregulated immune systems in hypertensive patients.[42] Accordingly, normalizing blood pressure in hypertensive patients can help avoid COVID-19 complications in these patients.
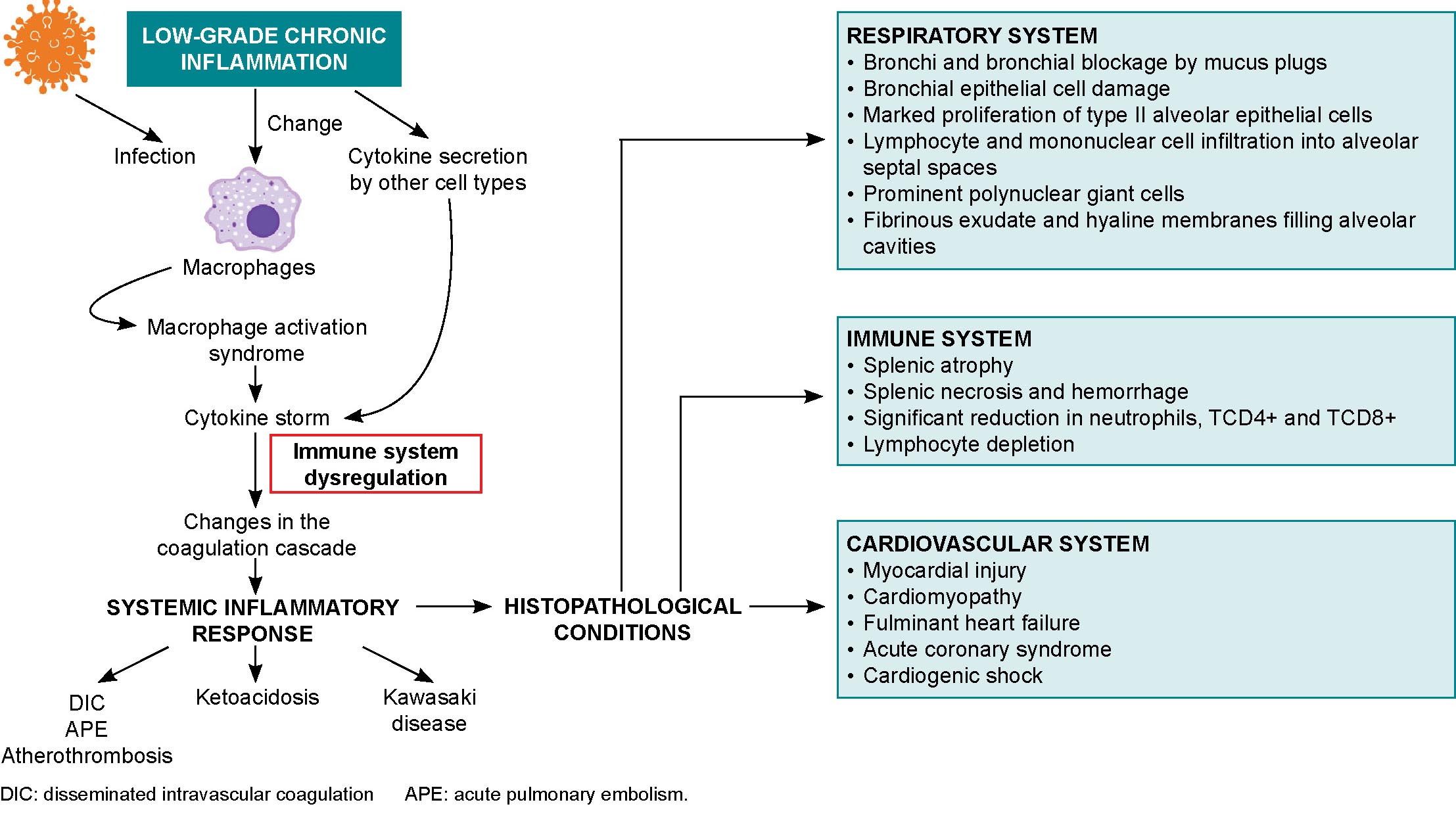
Possible mechanisms of myocardial injury in COVID-19 patients include entrance of the SARS-CoV-2 virus through myocardial ACE2 receptors, hypoxia, and cytokine storm-induced monocyte damage.[88] Cardiac consequences of SARS-CoV-2 infection range from cardiac arrhythmias and hypotension to acute coronary events and heart failure with cardiogenic shock.[42,88] Myocardial injury may be due to viral colonization, cytokine storms,[39] atherosclerotic plaque rupture, hypoxic injury, coronary spasms, formation of microthrombi, or direct vascular or endothelial injury.[88] Biopsies have shown low-grade myocardial inflammation[39] with interstitial edema[88] and viral particles in and around interstitial cytopathic macrophages but not in cardiomyocytes.[39] COVID-19 can trigger subclinical autoimmune myocarditis, and myocardial damage can cause a de novo autoimmune reaction (Figure 2).[42]
SARS-CoV-2 can destabilize or even shed atherosclerotic plaques during profound systemic inflammatory responses, cytokine storms, hemodynamic changes, or the polarization of immune cells toward more unstable phenotypes.[39,42] These processes, in conjunction with cytokine circulation in the heart and reduced oxygen supply, can cause coronary microvasculature dysfunction with inflammatory cardiomyopathy or atherothrombosis, leading to the acute coronary syndrome seen in deceased patients.[39]
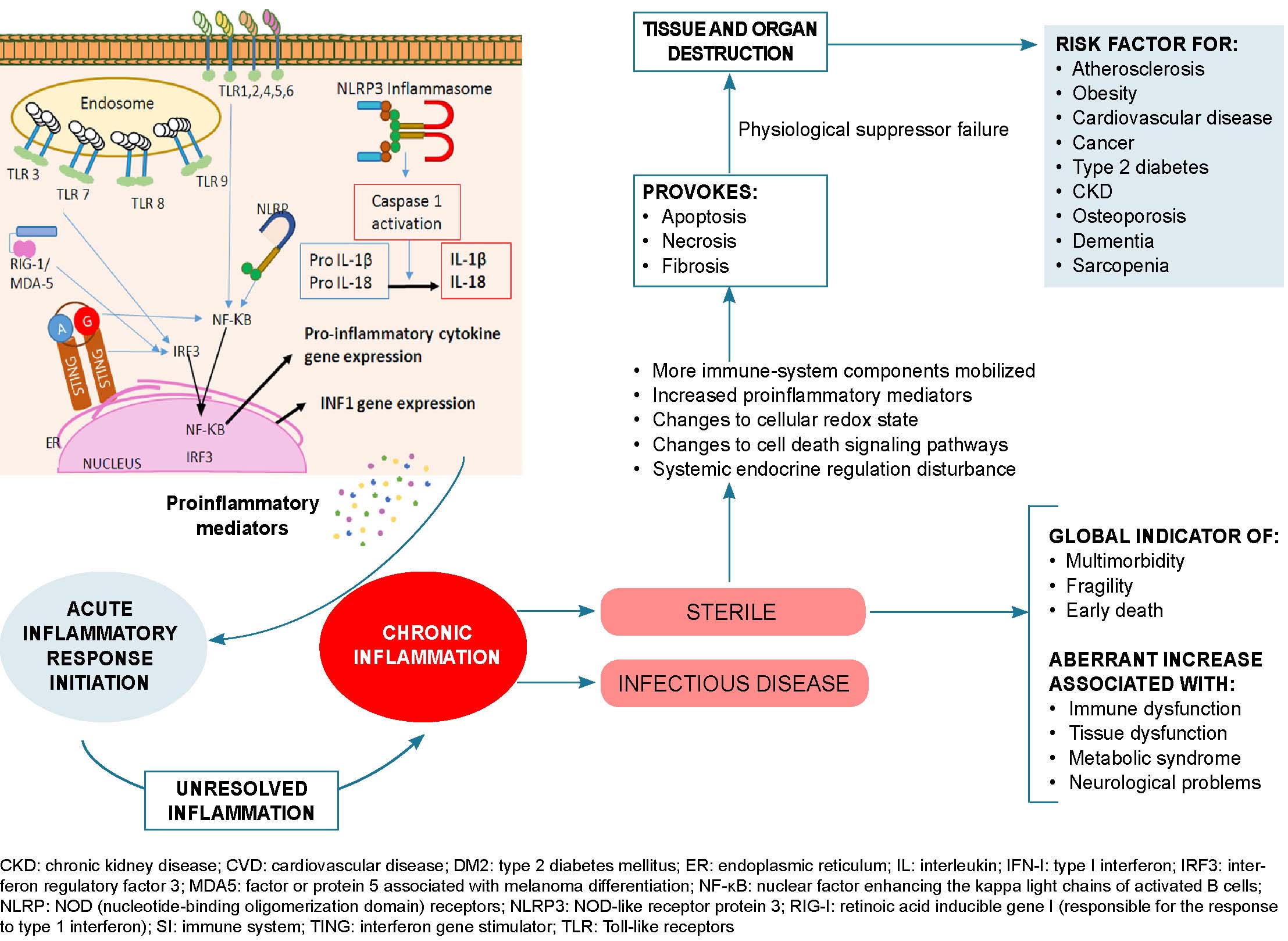
Oncoproliferative diseases Oncogenesis is an extremely complex process that involves immune, genetic and exogenous factors.[62] Rudolf Virchow first described the association between cancer and inflammation in 1863.[95] Inflammatory immune responses can be tumorigenic or antitumorigenic, depending on delicate balances between innate and adaptive immune system responses influenced by environmental and microenvironmental conditions.[96–98] A healthy, regulated immune response is considered antitumoral, while uncontrolled and excessive responses can induce chronic inflammation and proto-oncogenic conditions.[97] Low-grade inflammation associated with persistent infection may be carcinogenic.[96,99,100] Genetic and epigenetic factors also play a role in carcinogenesis.[101]
Chronic inflammation can trigger altered oncogene and tumor-suppressor gene expression.[96] This inflammation is associated with environmental factors, which, together with somatic mutations, are responsible for 90% of all cancers.[98] Chronic inflammation leads to structure loss and excessive tissue remodeling and DNA modification due to oxidative stress, increasing cancer risk.[101] Chronic inflammation is also implicated in obesity-related cancers like those of the liver and pancreas,[98,96] and obese patients are 1.6 times more likely to develop these cancers.[98] This inflammation is accompanied by elevated levels of insulin, glucose, leptin, C-reactive protein and IL-6, which can contribute to worse prognoses for overweight or obese COVID-19 patients.[99] Inflammation may also be involved in higher incidences of cancer in older adults. The aging microenvironment, notable within the tumor microenvironment, plays a key role in reprogramming tumor cells toward secretory phenotypes associated with senescence that have tumorigenic effects such as increased malignant phenotypes and tumor induction.[79]
Tumor-associated inflammation develops in tumorigenic microenvironments,[100] in which tumor-promoting immunity and antitumor immunity coexist.[101] In these microenvironments, there are elevated levels of inflammatory mediators, RNOS, microRNAs, and increased cyclooxygenase-2 activity that facilitate inflammation-mediated tumorigenesis.[96,99] The normal processes of cell proliferation, senescence and apoptosis are also affected, as are DNA mutation and methylation rates.[99] Proinflammatory mediators induce several molecular-signaling cascades that promote inflammatory states.[96] IL-23, secreted by Th17 lymphocytes, is a key factor in maintaining and expanding tumorigenic Th17 inflammatory cells, promoting inflammation, angiogenesis and reducing TCD8+ lymphocytes within the tumor microenvironment.[96] All these processes and factors have systemic repercussions, reduce the immune response to SARS-CoV-2 and favor uncontrolled inflammation that can lead to cytokine storms in already immunosuppressed patients.
Cancer patients are more susceptible to SARS-CoV-2 infection due to systemic immunosuppression caused by malignancy and treatment.[78] Cancer patients who have undergone major surgery, chemotherapy, radiation therapy and immunotherapy are at increased risk of contracting COVID-19, and its sequelae,[102] as are patients with lung, hematologic or metastatic cancers.[43,103] Anemia and hypoproteinemia have been found in COVID-19–positive cancer patients, a result of nutritional deterioration that negatively affects immunity and increases susceptibility to respiratory pathogens.[103] Lung cancer patients, who have impaired lung function and resistance, have high probabilities of developing severe anoxia and rapidly progressing to critical condition or death from COVID-19.[43,103] Chronic lung inflammation favors increased proinflammatory cytokines,[43] and poor immune response allows for viral spread, tissue destruction and progression to severe stages of the disease.[33]
Respiratory diseases COPD and asthma are the two most common and dangerous airway diseases.[104] Both are characterized by the presence of chronic inflammation.[105,106] These patients present with changes to lung anatomy and to the blood vessels that feed them.[88]
COPD Chronic obstructive pulmonary disease causes limitations in airflow and is associated with chronic airway and lung inflammation.[51] Other comorbidities are found in 60%–90% of COVID-19–positive COPD patients, aggravating symptoms and worsening disease prognosis.[37]
Defense barrier changes are observed in COPD pathology,[51] mainly failures in mucociliary mechanisms due to hair cell destruction, the presence of dehydrated mucus with changes to normal biophysical properties and mucin hypersecretion.[40] Low-grade inflammation is behind many of the changes caused by COPD, such as destruction of lung parenchyma and parenchymal vasculature, normal tissue repair disruption, small airway fibrosis and alveolar rupture.[51] This inflammation is characterized by an inadequate immune response with increases in neutrophils, macrophages, Th1 lymphocytes, B lymphocytes, vascular endothelial cadherins, platelet endothelial-cell adhesion molecules and E-selectin. These increases are associated with lung destruction and airflow limitation.[51] The protease/antiprotease relationship is altered, causing neurogenic damage and abnormalities in apoptotic, catabolic and senescent mechanisms. Imbalances between oxidizing and antioxidant agents causes activation of kinases and transcription factors, inflammatory mediator release, cell damage and apoptosis. Involved oxidants include reactive oxygen species (ROS) that damage cells, inactivate defense mechanisms, initiate inflammation and increase oxidative stress.[51]
COPD patients have increased ACE2 receptors in small airways,[107] but some authors argue that this may be mediated in the case of COVID-19 by the use of corticosteroid inhalants, which could limit lung damage in SARS-CoV-2 infection.[108] However, in COVID-19 animal models, ACE2 loss or deficiency is associated with lung injury,[35,109] and causes inflammation,[35] increased vascular permeability and acute respiratory distress syndrome.[109] Pre-existing endothelial damage to pulmonary capillaries is accentuated and leads to lung destruction, cardiovascular disease and cerebrovascular damage.[51] Pulmonary endothelial damage is considered the hallmark of acute respiratory distress syndrome.[38]
Asthma Bronchial asthma is a heterogenous disease characterized by chronic airway inflammation and variable remodeling, with a variety of clinical presentations and responses to treatment.[110] Most viral infections exacerbate asthma,[104,111,112] generally by inducing Th2-mediated responses, and high levels of proinflammatory cytokines and proinflammatory cytokine receptors.[111] Many of these reactions also appear during acute asthma attacks.
Several studies report that SARS-CoV-2 infection does not exacerbate asthma attacks,[111,113,114] and it is not associated with increased risks of hospitalization, severity or mortality, compared to patients without asthma.[114–116] Asthmatic patients generally have more comorbidities; however, greater severity and higher mortality levels have not been seen in COVID-19–positive asthmatics.[88,104,117] This may not be contradictory, as the course of COVID-19 in asthmatics depends on the persistence and severity of the patient’s asthma. To avoid or reduce risk of COVID-19 complications in asthmatics, it is important to control both the asthma and its inter-crisis treatment, which prevents structural and functional damage to the tracheobronchial tree.
Different asthma phenotypes show varying susceptibility to and severity of COVID-19.[116] In patients with type 2 asthma, we see chronic eosinophilic type 2 inflammation, the most common form of inflammation in asthma.[104,118] Type 2 asthmatics suffer from bronchial obstruction due to infiltration of innate immunity cells (primarily eosinophils, neutrophils, mast cells and macrophages)[104] and secretion of cytokines by innate immunity cells that shifts the immune response toward production of Th2 lymphocytes, type 2 B lymphocytes and allergen-specific IgE, which, in turn, perpetuates the inflammatory response.[106] Other mechanisms involved in persistent asthma are disruptions in inflammation resolution due to prolonged mast cell and eosinophil survival, decreases in lipoxin A4 in asthmatic airways, responsible for eosinophil apoptosis and for decreasing type 2 innate lymphoid cells (ILC2), and in NK cell activity.[119]
It has been hypothesized that the type 2 inflammatory response in asthmatics behaves differently than those of other pathologies,[10] and that it may act as a protective factor against COVID-19 contraction and progression,[120] due to Th2-biased immunity with cross-regulation between allergic immunity and altered interferon-mediated responses,[118] due to decreased production of bronchial epithelial cells and plasmacytoid dendritic cells.[106] The Th2-dominant environment is capable of regulating the late-phase hyperinflammation that typically marks severe respiratory viral diseases, but immunopathological processes are the hallmark of tissue damage.[118]
There are various schools of thought regarding ACE2 receptor involvement in SARS-CoV-2 infection in asthmatics. Calvielli suggests ACE2 expression is different in different types of asthma.[121] Other researchers suggest that respiratory epithelial cells in asthmatics show decreased ACE2 receptor gene expression,[120,122] and that this decrease overrides the minimal increase in transmembrane protease serine 2 (TMPRSS2) gene expression.[122] The reduced expression of the SARS-CoV-2 receptor could be due to the fact that IL-13 is involved in recruiting eosinophils in bronchial epithelia,[123] which can reduce ACE2 expression in human bronchial tissue (observed ex vivo).[120] ACE2 receptor expression is lower in patients with high allergic sensitization.[123,124] In contrast, patients with non-type 2 asthma have inflammatory profiles featuring Th1 and Th17 lymphocytes.[116,119] Non-type 2 asthmatics’ molecular phenotype is characterized by metabolic and mitochondrial pathways associated with the inflammasome and is generally accompanied by comorbidities like obesity, type 2 diabetes and hypertension. As they have low blood eosinophil numbers,[116] and ACE2 and TMPRSS2 gene expression levels similar to those in healthy people,[123,124] these asthmatics are susceptible to developing more severe forms of COVID-19.[116]
Scope, limitations and clinical implications of the review COVID-19 research generates such a volume of information that any review article runs the risk of missing important results. Many reviews only include articles written and published in English; this work also includes articles written in Spanish, but not in languages other than Spanish and English. This review focused on intersections of the immune system, low-grade inflammation and COVID-19 immunopathology with chronic diseases and age; other diseases that directly affect the immune system—such as immunodeficiencies or autoimmune diseases—were not included, nor were other chronic diseases such as kidney or neurogenerative diseases.
This review summarizes the main immunopathological alterations of the most frequent comorbidities described in COVID-19 patients, which directly influence the clinical course of the disease. This can aid patient management in clinical practice by identifying possible preventive actions designed to mitigate potential complications or progression to more severe manifestations of the disease.
FINAL CONSIDERATIONS
Low-grade chronic inflammation is characterized by a pro-inflammatory state with endothelial dysfunction and changes to the immune system, mainly in the innate immune response, with increases of proinflammatory mediators that generate pathogenic conditions and prevent elimination of the virus by triggering a dysregulated immune response. Inflammation is a common factor in non-communicable chronic diseases like obesity, type 2 diabetes, hypertension, cancer and COPD, all of which are risk factors for developing severe forms of COVID-19. Such risk is markedly increased when several of these factors occur in persons aged ≥60 years, except for those with type 2 bronchial asthma, where chronic eosinophilic inflammation protects against SARS-CoV-2 infection due to decreased interferon-mediated response and a reduction in ACE2 receptors. The links between immunosenescence, chronic disease and altered immune response in COVID-19 deserves continued attention by researchers.