INTRODUCTION
Parkinson’s disease (PD) is the second most common aging-related neurodegenerative disease after Alzheimer’s disease.[1] The only prevalence estimate available for Cuba is that for an urban area of Havana (135 per 100,000 population aged ≥15 years), suggestive of its magnitude as a health problem.[2] Epidemiological research in other countries has yielded widely varying rates, reflecting differences in study design, variables such as age and sex, and data analysis techniques, with door-to-door studies the most reliable. A recent evidence review of PD etiology and prevalence showed differences by geographic zone and rising prevalence with aging.[3] For example, researchers in Brazil found a prevalence of 7200 per 100,000 population aged >64 years, rising to 14,300 per 100,000 population aged ≥85 years.[4] Overall, it is estimated that close to 5 million people have PD worldwide.[5]
Currently, PD drug therapy is based on dopamine precursors and agonists.[6] These drugs control symptoms during the disease’s early stages,[7] but as the disease progresses, emerging symptoms do not respond to dopaminergic stimulation.[8] The resulting loss of dexterity and independent gait plus cognitive decline become disabling. This situation has spurred the search for second-line neuroprotective strategies to slow or delay neurodegeneration.[9]
One potential neuroprotectant is erythropoietin (EPO), a cytokine known to be a hematopoietic growth factor that plays an important role in control of tissue oxygenation.[10] It was initially thought that this was its only function; however, like other members of the cytokine superfamily, it has other functions, such as neuroprotection through an antiapoptotic effect, as an antioxidant and by preventing cell damage from the nitric oxide cascade─in other words, against mechanisms involved in neuronal death.[11] In addition, it has neurotrophic and neurogenic effects.[12]
In 1985, recombinant human erythropoietin (rHuEPO) began to be used in end-stage renal disease patients. In 1989, it was approved by the US Food and Drug Administration. At present, it is widely used to treat anemias related to premature birth, kidney failure, cancer, chronic inflammatory diseases and HIV infection.[13,14]
The neuroprotective effect of rHuEPO in humans has been demonstrated by positive results in patients with neurological disorders such as cerebrovascular disease,[15] Friedreich’s ataxia,[16,17] multiple sclerosis[18] and schizophrenia.[19] Surprisingly, the only report of its use in PD patients is an incidental observation in a multiple sclerosis study that included two PD patients as controls, in whom it was well tolerated.[18] Even though rHuEPO has been a strong candidate as a neuroprotectant, prompting numerous preclinical studies with positive findings, until now no study in humans has been published on its safety, tolerability or efficacy for PD, according to a recent review.[20]
Cuba produces rHuEPO (ior-EPOCIM, Molecular Immunology Center), which is registered and approved for use in humans in Cuba and in a number of other countries. For some time it has been used effectively in managing EPO-deficiency anemia.[21,22]
The foregoing led us to propose a preliminary exploratory study (proof of concept) to assess the safety and possible neuroprotective effect of Cuban rHuEPO (ior-EPOCIM) in PD patients.
METHODS
Study design An exploratory study (proof of concept) was conducted to assess the safety of ior-EPOCIM in PD patients at a dose reported safe in patients with renal disorders,[23] and secondarily, to evaluate its possible neuroprotective effect.
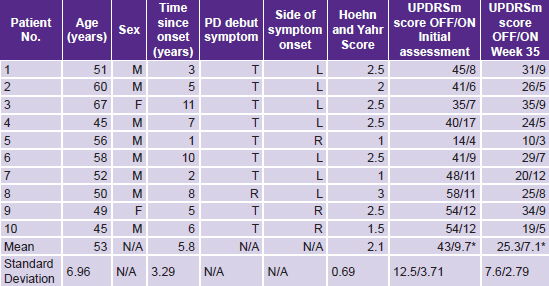
The universe comprised patients seen at the International Neurological Restoration Center (CIREN, the Spanish acronym) outpatient Movement and Neurodegenerative Disorders Clinic from May through July 2008. Of a total of 27 patients interviewed who fulfilled the London Brain Bank’s operational criteria for PD[24] and the other inclusion criteria listed below, it was only possible to include 10 (8 men, 2 women), aged 47–65 years.
Seven had symptom onset in the left half of the body and disease severity up to stage 3 on the Hoehn and Yahr scale (indicator of PD stage and severity: least severe 1, most severe 5).[25] Stage 4 and 5 patients were excluded because they were very disabled and unable to participate as ambulatory patients in a lengthy study.
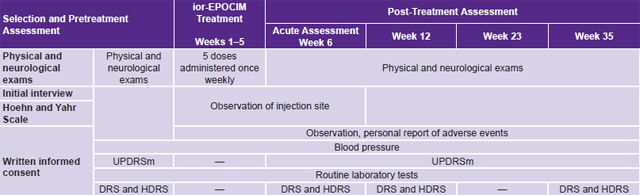
Pretreatment selection and assessment took place from May through July 2008. Treatment phase lasted five weeks in August and September 2008; and assessments were made in weeks 6, 12, 23 and 35. Table 1 provides details.
Inclusion criteria Patients aged 45–75 years, willing to participate in the study; ≥1 year since disease onset; good response to antiparkinsonian treatment with levodopa (>30% change in motor score on the motor section of the Unified Parkinson’s Disease Rating Scale, (UPDRSm); acceptable overall health; no prior polycythemia (hematocrit ≤50%); and no significant cognitive decline as measured by clinical assessment, DRS (Dementia Rating Scale) and the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV).[26]
Exclusion criteria Refusal to participate; cognitive decline; known hypersensitivity to products derived from eukaryotes or hypersensitivity to human albumin; pregnancy or breastfeeding; hypertension; immunosuppressant, androgen or anabolic steroid treatment in the month prior to recruitment; sepsis or active infection; active acute or chronic inflammatory diseases; hematologic diseases, such as sickle cell disease, myelodysplastic syndromes, active clotting or bleeding disorders; malignant tumor or cancer treatment; or alcoholism or drug addiction in the two years prior to inclusion assessment.
Discontinuation criteria Appearance of an exclusion criterion (in the case of an isolated increase in blood pressure, the principal investigator had discretion to keep a patient in the study if response to treatment was satisfactory); a potentially life-threatening adverse effect in the judgment of the principal investigator; any psychotic decompensation; accidents or other diseases during the therapy period.
Ethical considerations Patients and accompanying family members provided written informed consent. Information provided included the selection process, treatment, monitoring, and publication of data. The CIREN ethics committee approved the study after scientific, methodological and ethical review.
Treatment The rHuEPO used was ior-EPOCIM (Molecular Immunology Center, Cuba) in liquid form in 10,000 IU (1 mL) vials, whose active principle is the 165-amino acid glycoprotein with a molecular weight of 34,000. It was administered by subcutaneous injection in the deltoid region. A dose of 60 IU/kg of body weight (2500–5000 IU per injection, depending on weight), equivalent to doses used in renal disorders, was chosen for its known safety.[23]
Table 1: Proof-of-concept study timeline for ior-EPOCIM in Parkinson’s disease patients
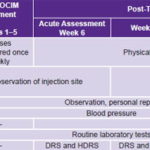
DRS: Dementia Rating Scale HDRS: Hamilton Depression Rating Scale Routine laboratory tests: Hemoglobin, hematocrit, white blood cell count, erythrocyte count, mean cell volume, mean cell hemoglobin, platelet count, erythrocyte sedimentation rate UPDRSm: Unified Parkinson’s Disease Rating Scale–motor exam (the only assessment done both ON and OFF antiparkinsonian medication)
Treatment was ambulatory and provided at CIREN’s Movement and Neurodegenerative Disorders Clinic. Personnel accredited to work on the study protocol administered the indicated dose once a week for five consecutive weeks (Table 1). To avoid biasing results, antiparkinsonian drug treatment prescribed by the attending neurologist was maintained during the study, with no new drugs prescribed.
Since measuring baseline motor function was essential, antiparkinsonian drugs were withdrawn at determined intervals to administer the UPDRSm in two conditions, ON and OFF:[27] ON when the patient was under the effects of antiparkinsonian drugs and hence had optimal mobility and mental status, and OFF when the patient had gone for a period without dopaminergic stimulation and parkinsonian symptoms hence became evident.
For the study, antiparkinsonian drugs were withdrawn 12 hours before administering UPDRSm (OFF condition), as can be seen in the timetable (Table 1); following the exam, medication was resumed to assess the ON condition the same day. Except for UPDRSm in OFF time, all other evaluations were done in the ON condition.
Safety Assessment was based on adverse events (AE), defined as any event clinically harmful to the patient, whether or not it was related to the product under study. AEs were classified according to their intensity: mild (effects well tolerated by patient, causing minimum discomfort, not interfering with daily activities), moderate (discomfort sufficient to interfere with daily activities), and severe (effects impeding daily activities or potentially life threatening).[28] Changes in hemoglobin and other hematologic parameters and an increase in blood pressure were considered potential adverse effects, because the product is a hematopoietic growth factor administered in this case to patients without anemia.[29]
Hematologic assessment. The following tests were performed (range of normal values in parentheses): hemoglobin concentration (men 13–17 g/dL; women 12–16); hematocrit (40–50%); white blood cell count (5–10 x10³/μL); total erythrocyte count (4.5–5.5 x 106/μL); mean cell volume (82–92 fL); mean cell hemoglobin (26–32 pg/cell); mean cell hemoglobin concentration (300–360 g/dL); platelet count (150–350 x10³/μL); and erythrocyte sedimentation rate (men 0–10 mm/hr; women 0–20 mm/hr).
Blood testing was carried out in CIREN’s clinical laboratory (ISO 9000-certified), using the Sysmex Hematology Analyzer (model KX-21N, Japan).
Blood pressure assessment. Systolic and diastolic blood pressure were measured once in a sitting position. Hypertension was defined by systolic pressure of ≥140 mm Hg and/or diastolic pressure of ≥90 mm Hg, according to European Society of Hypertension standards.[30]
Evaluation of possible neuroprotection We explored variables related to motor function (cardinal symptoms of PD), as well as patients’ baseline (pretreatment) and post-treatment neuropsychological and emotional status.
Neurological assessment. Motor impairment was assessed using the UPDRSm (Unified Parkinson’s Disease Rating Scale, motor examination),[27] which enables recognition of motor dysfunction. UPDRSm comprises the following domains: speech, facial expression, tremor at rest, action or posture tremor, axial and limb rigidity, rapid succession and rapidly alternating hand movements, leg agility, arising from a chair, posture, gait, postural stability, and body bradykinesia and hypokinesia. Each item is scored on a scale ranging from 0 (normal or absence of symptoms) to 4 (maximum impairment). This was the only technique or procedure used on patients both with and without the dopaminergic drug prescribed prior to entering the study (ON and OFF conditions, respectively).
Cognitive assessment. Degree of cognitive decline was measured by the Dementia Rating Scale (DRS),[31] commonly used for that purpose in PD. The scale covers five domains: attention, initiation–perseveration, construction, conceptualization, and memory, with a score ranging from 0–144, where normal is a score of ≥137; mild dementia, 111–136; moderate dementia, 82–110; severe dementia, 66–81; and profound dementia, ≤65.
Emotional status assessment. The Hamilton Depression Rating Scale (HDRS)[32] was used to classify and interpret depression severity. Scores range from 0 to 40 with the following categories: no depression, 0–9; mild depressive symptoms, 10–13; moderate depression, 14–17; and severe depression, ≥18.
Data collection and analysis Patients’ clinical history data were stored and tabulated in an Excel database and later exported for processing with statistical software, S-PLUS 8 for Windows (2007). Safety was assessed by qualitative description of adverse events occurring; numerical values were summarized using arithmetic mean and standard deviation.
All statistical analyses were done using a linear mixed-effects model[33] in which values from all measurements were entered for each variable before, during and after treatment, as was initially designed.
A preliminary exploratory analysis was done to assess significant changes over time between initial and post-treatment assessments, using a fixed-effects linear model applied successively to scores for variables on the UPDRSm (OFF and ON conditions), DRS and HDRS scales.
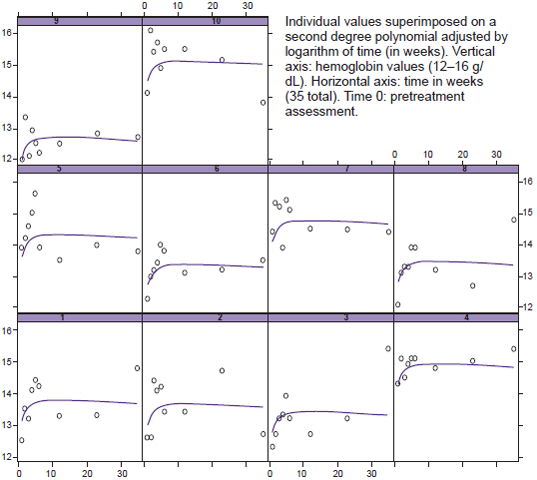
Hemoglobin behavior through treatment and post-treatment stages was analyzed by applying a quadratic polynomial model (quadratic mixed-effects model) using duration (log of week number) as the dependent variable.
Finally, changes in UPDRSm in OFF and ON conditions were assessed taking into consideration potential confounders—DRS, Hamilton, age, PD severity stage (Hoehn and Yahr) and time elapsed since disease onset—to see if positive effects found were statistically independent of these.
Graphs were produced to display fixed-effects adjusted functions in each patient over time.
RESULTS
Patient adherence to the study protocol was 100%.
There were mild adverse events in three patients during the course of the study, all reported during the treatment phase. There were no severe or life-threatening adverse events, nor any requiring hospitalization. There were no local reactions at the injection site. One patient experienced precordial discomfort and high blood pressure readings following the third injection; both were controlled with symptomatic medication and did not recur during the remainder of the study. This patient had previously experienced hypertensive events but did not mention them in the recruitment interview, since he was not on antihypertensive treatment; it would have been grounds for exclusion (with the exception of this case, patient blood pressure readings remained within normal limits). A second patient had leg fatigue one or two days following each injection; it was deemed mild enough not to require treatment and resolved spontaneously. A third patient with a history of renal colic had an episode following the third injection; it resolved satisfactorily with the usual medication. There were no new adverse events in the post-treatment phase.
Among hematologic variables, only hemoglobin changed with treatment. At the beginning of the study, mean hemoglobin concentration for the group was 13 g/dL and six patients had levels of < 13 g/dL. Hemoglobin concentration increased over baseline levels in all patients; changes were statistically significant (p < 0.001). However, although there was an increase at the start of treatment, levels tended to stabilize in the post-treatment phase. Even though in four cases (patients 1, 3, 4, and 8) final hemoglobin was far above the individuals’ initial levels, it remained below the upper limit for normal, so phlebotomy was unnecessary (Figure 1).
Other unexpected reactions that were considered beneficial were moderately increased appetite in one patient starting with the first dose—resulting in a 5-kg weight increase by week 35—and mood improvement reported by four patients.
Figure 1: Changes in individual hemoglobin concentration

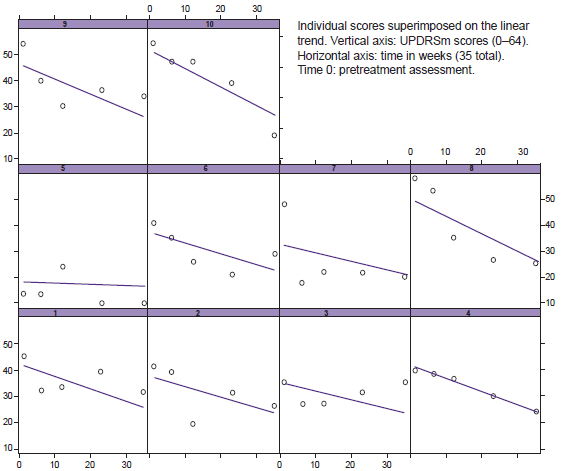
During drug withdrawal (OFF condition), total UPDRSm scores decreased in weeks 6, 12, 23 and 35 compared with baseline or pretreatment status (Figure 2). The difference observed between mean group score for the OFF condition before treatment (43) and at week 35 (25.3) was highly statistically significant (p < 0.001), as were positive changes in individual motor function scores (Table 2). No significant changes were found in the ON condition.
Individual UPDRSm elements showing beneficial and statistically significant clinical changes in the OFF condition were facial expression, gait hypokinesia, tremor at rest of the left arm and both legs, rigidity of the left leg, and axial rigidity. Scores for all decreased between the first and final assessments. Other parameters—such as gait, speech, hypokinesia on opening the left hand, and posture—showed significant changes only late in the post-treatment period (week 23 or 35).
Additional statistical analysis to assess effects on UPDRSm score in OFF of other variables studied, such as study duration (35 weeks), DRS and HDRS scores, age, disease severity, and time elapsed since onset of disease, showed that only post-treatment time was highly significant (p < 0.001) for motor function. A less powerful influence was seen for increase in age and time since onset of disease in years (also statistically significant, p = 0.05), which could indicate a tendency for younger patients who have had the disease for less time to have lower (more favorable) scores on UPDRSm in the OFF condition post treatment (Table 2).
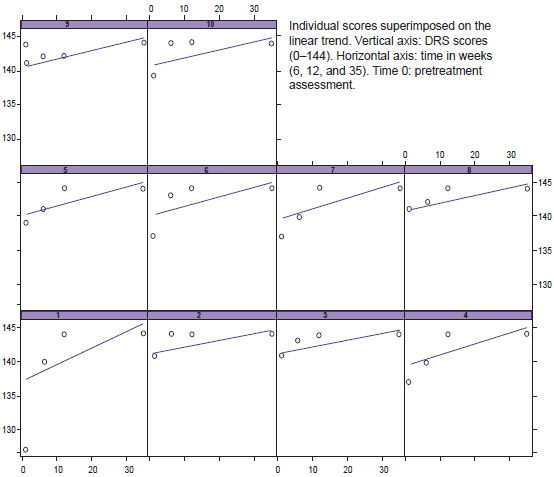
Total DRS score showed a favorable and significant increase in all subjects over baseline status (p < 0.001) (Figure 3), with the attention subtest contributing to this change (p = 0.013).
Before starting treatment, eight of the ten subjects were severely depressed (group mean HDRS score 23.4 points). HDRS scores dropped to normal (group mean 12.1) for all but three patients (1, 8, and 9). The effect was statistically significant (p = 0.013) and was consistent with improved mood reported by four patients.
DISCUSSION
The adverse effects of treatment with ior-EPOCIM found in this study were mild and occurred in few patients, consistent with its reported safety in renal disease and cancer patients.[21–23] Unlike in these studies, our patients were not anemic. Increases in hemoglobin in all patients were expected, given that ior-EPOCIM is hematopoietic (its main indication) and our findings are similar to those reported in studies of rHuEPO as a neuroprotectant in nervous system disorders.[15–19] Phlebotomy is frequently required during treatment with rHuEPO to bring hemoglobin values down to normal,[16–19] but was not necessary in our study because in no case did hemoglobin values exceed the upper limit of sex-specific norms.
The difference observed between our study and others is closely related to study dose. The ataxia study[16,17] used a dose of 15,000 IU per week (three or more times the dose received by participants in this study), resulting in increases in hematologic parameters and need for phlebotomy in half the patients studied, leading researchers to reduce rHuEPO dose to 6000 IU per week. Another factor that may have contributed to these results is that the majority of our patients had initial hemoglobin concentrations and hematocrits close to lower normal limits, and thus increases produced by ior-EPOCIM treatment can in fact be considered beneficial.
Increased blood pressure in one patient with a history of hypertension could be related to the product; as has been reported in its use for anemia.[13] Likewise, leg fatigue was reported in the clinical trial for multiple sclerosis.[18] The one adverse event that did not appear to have a causal relationship with product use was renal colic, observed in one patient with a long history of such events. The patient’s crisis following the third dose of ior-EPOCIM followed the same course as previous ones. We found no reports of renal colic in other studies of rHuEPO.
The authors of the above-mentioned ataxia study also observed improvements in overall wellbeing with rHuEPO,[16] as reported by four of our patients.
Since changes in antiparkinsonian medication prescribed during a study can be an important confounder, clinical trials involving PD patients maintain the original prescription; furthermore, its withdrawal causes a disabling functional decline. This is most important in long-term and ambulatory studies such as ours, because drug withdrawal interferes with basic survival, since motor symptoms compromise gait, speech, swallowing, and other neurovegetative functions. Hence, with one exception, all assessments were conducted in the ON status. However, to assess baseline motor conditions and evaluate the possible neuroprotective effect of ior-EPOCIM, it was necessary to withdraw antiparkinsonian medication for brief intervals and administer UPDRSm in ON and OFF treatment conditions. Antiparkinsonian medication was withdrawn 12 hours before administering the UPDRSm (OFF condition), immediately resuming the prescribed medication to assess motor function in the ON condition the same day.
Figure 2: Variations in individual UPDRSm scores in OFF condition from baseline to study completion

Table 2: Study participants’ demographic and clinical data; and motor function before and after ior-EPOCIM treatment for Parkinson’s disease
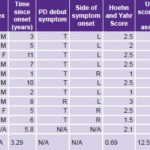
UPDRSm: Unified Parkinson’s Disease Rating Scale–motor exam T: Tremor R: Rigidity L: Left R: Right OFF: Anti-parkinsonian medication suspended 12 hours before motor assessment ON: Patient on anti-parkinsonian medication at time of motor assessment * p < 0.001
The clinically positive and statistically significant changes in UPDRSm scores (OFF condition) from baseline status to post-treatment assessments were quite striking. The improvement was seen in reduced scores for UPDRSm subsection items on gait hypokinesia, left arm tremor at rest, tremor in both legs at rest, and left arm rigidity. These findings were interpreted as the result of therapeutic action by ior-EPOCIM on motor status during antiparkinsonian medication withdrawal. The effect was not symmetrical: the side that benefited most was the one presenting the most parkinsonian symptoms, in this case the left, where symptom onset occurred in seven of the ten patients. The fact that these changes in UPDRSm scores were more evident in the OFF condition appears to demonstrate the mechanisms through which EPO protects dopaminergic neurons.[34]
Figure 3: Variations in individual DRS scores over time

The response of this neuronal population is not only conditioned by striatal dopamine concentration;[35] in fact, it is frequently observed that treatments affecting dopaminergic transmission produce a “ceiling” effect with saturation of neuroreceptors. Furthermore, agents that have an impact on denervation of basal ganglia or abnormal dopaminergic activity frequently diminish intensity of motor manifestations in the OFF condition; i.e., without dopaminergic medication (“threshold effect”).[36]
DRS[31] was used not only to rule out dementia during preliminary assessment, but also to help assess ior-EPOCIM’s effects on overall cognitive status. Surprisingly, cognitive function did improve, demonstrated by an increase in DRS scores, especially for the attention subscale. This result coincides with those of other studies (specifically in anemia and renal diseases) reporting beneficial effects of rHuEPO on cognition.[37,38] Findings from animal models and human studies suggest that this cognitive improvement may be secondary to various mechanisms, such as kinase activation by second messengers that stimulate dendritic proliferation.[20,39]
We may surmise a neuroprotective effect from our findings, especially regarding attention, since this is independent of the probable effects of memory and learning.
A problem in interpreting motor and cognitive effects—and a limitation of our study—is the lack of a control group to rule out placebo effect. An improvement in mood was also seen, demonstrated by the significant decrease in HDRS score, and we cannot rule out the possibility that emotional state influenced the positive results observed in motor function and cognitive status.
Support for such a hypothesis is found in results from a group of PD patients who served as controls for another group submitted to subthalamotomy. The PD controls derived motor and emotional benefits from the specialized attention involved in participating in a research protocol.[40]
However, we cannot attribute improvements observed in this study entirely to placebo effect, since they persisted for 30 weeks following rHuEPO therapy, time considered sufficient to rule out placebo effect.[41] Additionally, statistical analysis, using mixed-effects linear analysis and removing the effect of confounders, found no interference from cognitive and emotional scores on the positive motor changes reported.
The small number of participants also limits the study’s power to infer a neuroprotective effect from responses observed. Though preliminary, these results are encouraging and fulfilled the objectives of an exploratory proof-of-concept study. The ior-EPOCIM dose administered was safe for these patients and the positive effects observed could suggest its potential effectiveness as a treatment for PD.
This study is a first step toward implementation of a Phase I clinical trial to be conducted by CIREN, which in turn is expected to lead to new studies of ior-EPOCIM in Cuba to answer the outstanding questions. The current study provides baseline information on dose and length of use in future studies.
CONCLUSIONS
Our research found ior-EPOCIM to be safe for PD patients at the dose and length of treatment used. Preliminary positive responses in motor, cognitive and affective domains suggest potential neuroprotective effects of the product that should be tested in clinical trials designed with this objective, such as those currently in development at CIREN.